 гІгУСьгђ
гІгУСьгђ
EEMзлЪіЃКВФСЯЛљвђзщЗНЗЈгаЖрЧПДѓЃППДDFTМЦЫудкШЋЙЬЬЌЕчГижаДгЩшМЦЕНКЯГЩЕФИпаЇгІгУ
QQбЇЪѕНЛСїШКЃК1092348845

ЯъЯИНщЩм
ЁОбаОПБГОАЁП
МюН№ЪєРызгЕчГиБЛЙуЗКгІгУгкБуаЏЪНЕчзгЩшБИКЭЕчЖЏЦћГЕжаЃЌФПЧАжЦдММюН№ЪєРызгЕчГигІгУЕФЦПОБЮЪЬтЪЧЧБдкЕФАВШЋвўЛМЃЌгыДЋЭГЕчГижаЪЙгУЕФвзШМЁЂвзБЌЕФвКЬЌгаЛњЕчНтжЪЯЂЯЂгаЙиЁЃбаЗЂвдЮоЛњРрЙЬЬЌЕчНтжЪ(SSE)ЮЊЛљДЁЕФШЋЙЬЬЌЕчГиЪЧНтОіЧБдкЕчГиАВШЋадЮЪЬтЕФгааЇЭООЖЁЃФПЧАЙЬЬЌЕчНтжЪЬхЯЕДѓжТЗжЮЊСНДѓРрЃКбѕЛЏЮяЕчНтжЪЁЂСђЛЏЮяЕчНтжЪЁЃбѕЛЏЮяЕчНтжЪОпгаНЯКУЕФЮШЖЈадЁЂЖдПеЦјКЭЫЎЗжВЛУєИаЁЂЭЌЪБОпгаНЯПэЕФЕчЛЏбЇДАПкЕШгХЪЦЁЃЕЋвВДцдкРызгЕчЕМТЪНЯЕЭЁЂОЇНч/ОЇСЃЕчзшНЯЁЂвзДрЖЯЁЂжЦБИЩеНсЮТЖШНЯИпЕШЮЪЬтЁЃСђЛЏЮяЕчНтжЪОпБИГЌИпЕФРызгЕчЕМТЪЁЂКУЕФбгеЙадЁЂаЁЕФОЇНч/ОЇСЃЕчзшЁЂНЯЕЭЕФжЦБИЩеНсЮТЖШЕШЬиеїЁЃЕЋЦфЖЬАхЭЌбљУїЯдЃЌР§ШчЃКЕчбЙДАПкеЁЂгыИпбЙе§МЋВФСЯМцШнадВюЁЂгыН№ЪєLiИКМЋЮШЖЈаддуИтЁЂЖдПеЦјКЭЪЊЖШУєИаЕШЁЃЬНбАзлКЯадФмИќМггХвьЕФаТаЭПьРызгЕМЬхГЩЮЊиНашНтОіЕФЙиМќПЮЬтжЎвЛЁЃДЫЭтЃЌШЋЙЬЬЌЕчГижаЕФЙЬ-ЙЬНчУцЕФЮШЖЈадЁЂМцШнадЁЂЦЅХфЖШЕШжюЖрЮЪЬтШдШЛбЯжижЦдМзХШЋЙЬЬЌЕчГиЕФЕчЛЏбЇадФмЁЃ
ДгХгДѓЕФЪ§ОнПтжаЃЌПМТЧЮоЧюЕФЛЏбЇГЩЗжзщКЯЃЌбАеваТаЭЙЬЬЌЕчНтжЪЕФЬхЯЕЃЌНіНіЭЈЙ§ЪЕбщЪдДэКмФбПьЫйОЋзМЕибАевЕНадФмгХвьЕФЕчНтжЪХфЗНЁЃСэЭтвЛЗНУцЃЌеыЖдШЋЙЬЬЌЕчГижаЙЬ-ЙЬНчУцЮЪЬтЃЌашвЊАКЙѓЕФдЮЛВтЪдВХФмБцЮіЧхГўЃЌНіЭЈЙ§жїЙлвмЖЯКмФбАбЮеЙиМќадПЦбЇЮЪЬтЁЃвђДЫЃЌиНашвЛЬзПЭЙлЕФбаОПЗНЗЈЃЌМгЫйаТаЭЕчНтжЪЕФЬНбАЙЄзїЃЌВЂЧвЮЊЙЙжўвЛЬхЛЏШЋЙЬЬЌЕчГиЬсЙЉКЯРэЛЏЗНАИЁЃжЃжнДѓбЇЩлЙњЪЄНЬЪкПЮЬтзщЃЌЭЈЙ§ећКЯВФСЯЛљвђзщЗНЗЈЃЌеыЖдЙЬЬЌЕчГижаЕФЙиМќЮЪЬтЃЌаЮГЩСЫвЛећЬзЕФDFTЫуЗЈЃЌНјааИпЭЈСПМЦЫуЃЌЬНбАаТаЭЙЬЬЌЕчНтжЪвдМАЬсЙЉНтОіШЋЙЬЬЌЕчГиЕФЙЬЙЬНчУцМцШнадЕФКЯРэадЗНАИЃЌЮЊКѓајЪЕбщбаЗЂЬсЙЉСЫгаСІЕФРэТлжЇГХЁЃ
ЁОГЩЙћМђНщЁП
жЃжнДѓбЇЩлЙњЪЄНЬЪкбаОПЭХЖгдкEnergy Environ. MaterЩЯЗЂБэЬтЮЊЁАFirst Principle Material Genome Approach for All SolidЉ\State BatteriesЁБЕФЮФеТЁЃ
ЁОЭМЮФЕМЖСЁП
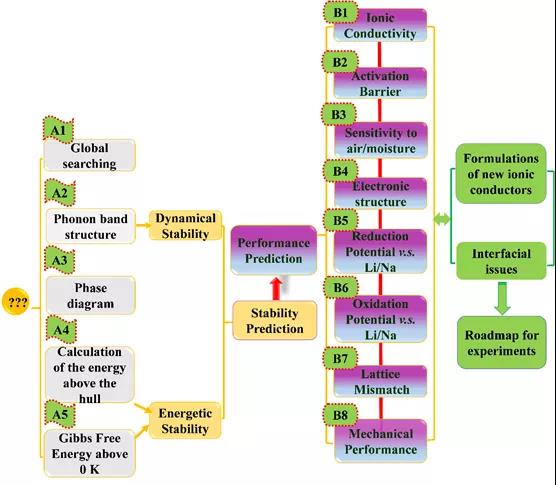
ЭМ 1. ВФСЯЛљвђзщЗНЗЈСїГЬЁЃ
ИпЭЈСПВФСЯЛљвђзщЗНЗЈМЦЫуСїГЬЃЌШчЭМ1ЫљЪОЃЌАќРЈЃЌЃЈAЃЉШШСІбЇКЭЖЏСІбЇЮШЖЈаддЄВтЃЌЃЈBЃЉЙЬЬЌЕчНтжЪЙиМќадФмдЄВтЃЌСНДѓВПЗжЁЃ(A)ВПЗжКИЧЕФВФСЯЛљвђзщЗНЗЈАќРЈЃКUSPEXвХДЋЫуЗЈЃЌЩљзгДјМЦЫуЃЌЯрЭМЗжЮіЃЌ0 KаЮГЩФмМЦЫуЃЌКЌЮТМЊВМЫЙздгЩФмМЦЫуЁЃЖдФтЩшМЦВФСЯЕФНсЙЙЮШЖЈадКЭШШСІбЇЮШЖЈадГфЗждЄЙРЃЌБЃеЯЫљЩшМЦВФСЯдкКѓајЙЄзїжаЕФПЩЪЕЪЉадЁЃ(B)ВПЗжЃЌЖдАќРЈРызгЕчЕМТЪЁЂРЉЩЂМЄЛюФмЁЂЕчзгНсЙЙЁЂбѕЛЏЛЙдЕчЮЛЁЂОЇИёЪЇХфЖШЁЂСІбЇадФмЕШЙиМќадФмНјааПЭЙлдЄВтЃЌИпЭЈСПЬНбАаТаЭЙЬЬЌЕчНтжЪЁЂВЂеыЖдЙЬЬЌЕчГижаЕФЙЬ-ЙЬНчУцЮЪЬтЃЌЬсИпКЯРэЛЏЗНАИКЭРэТлжЇГХЁЃ
СђвјерПѓаЭLi6PA5XЕчНтжЪЕФРэТлЩшМЦКЭЪЕбщжЦБИЃК
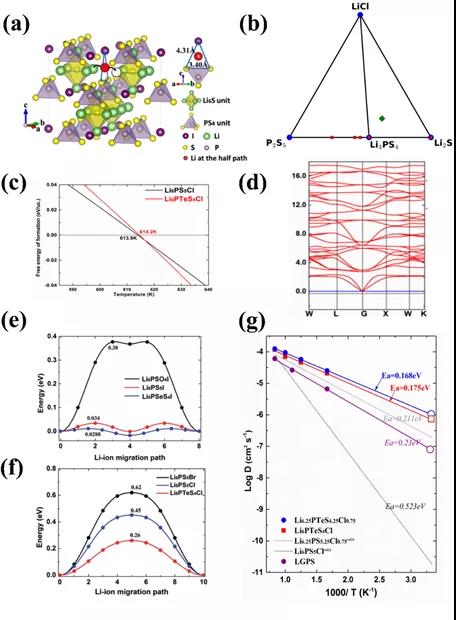
ЭМ2. (a)Li6PS5XжаЕФLi+РызгРЉЩЂЭЈЕРгыЪфдЫЦПОБЁЃ(b) Li6PS5ClЕФРэТлЯрЭМЁЃ(c)ЭЈЙ§МЊВМЫЙздгЩФмМЦЫуЕУГіЯрзЊБфЮТЖШЁЃЃЈdЃЉЭЈЙ§ЩљзгДјМЦЫудЄВтLi6PTeS4ClЕФНсЙЙЮШЖЈЁЃ(e)ЁЂЃЈfЃЉРћгУCI-NEBМЦЫуЗНЗЈЃЌУїШЗИФадЗНАИгыяЎРызгРЉЩЂЕФЖдгІЙиЯЕЁЃЃЈgЃЉЭЈЙ§AIMDМЦЫудЄЙРяЎРызгЪфдЫФмСІЁЃ
СђвјерПѓаЭLi6PS5XЕчНтжЪЙЙаЭжаЃЌДцдкS-X-SШ§НЧаЮЕФЦПОБЃЌЃЈШчЭМ2aЫљЪОЃЉЃЌМЋДѓЕФЯожЦзХяЎРызгЕФПьЫйЪфдЫЁЃЙЪЃЌиНашЭЈЙ§ИФадЃЌДђЭЈРызгЪфдЫЦПОБЃЌЪЧЪЕЯжЦфяЎРызгЕФПьЫйЪфдЫФмСІЕФЙиМќЁЃБОВПЗжЙЄзїеыЖдСђвјерПѓаЭЕчНтжЪЃЌЬсЙЉКЯРэЛЏЗНАИЁЃВЂЖдЦфЮШЖЈадКЭРызгЪфдЫФмСІзіГіГфЗждЄЙРЁЃОпЬхШчЯТЃКЭМ2ЃЈbЃЉЃЌЯрЭМЗжЮіЃЌВЮееLi3PS4КЭLiCl, Li6PS5ClЮЊбЧЮШЯрЃЛЭЈЙ§МЊВМЫЙздгЩФмМЦЫуЃЈЭМ2ЃЈcЃЉЃЉЃЌЗЂЯжЕБЮТЖШИпгк614KЪБЃЌLi6PS5ClЛсзЊБфГЩзюЮШЯрЃЌЮЊКѓајЪЕбщЙЄзїЬсЙЉШШДІРэЗНАИЁЃЭЈЙ§ЩљзгДјМЦЫуЃЈЭМ2ЃЈdЃЉЃЉ,ЮоащЦЕЫЕУїЦфНсЙЙЮШЖЈЃЌШєВЛЮШЖЈдђЫЕУїЫљЩшМЦНсЙЙВЛЮШЖЈЃЌЛсЗЂЩњЯрзЊБфЁЃеыЖдЕчНтжЪЕФЙиМќРызгЪфдЫадФмЃЌЃЈЭМ2ЃЈeЃЉ-(g)ЃЉ,CINEBгыAIMDМЦЫуЯрНсКЯЃЌБцЮіВЂгХбЁзюМбИФадЗНАИЁЃЭЈЙ§БШЖдРызгЕчЕМТЪКЭРЉЩЂМЄЛюФмЃЌгХбЁГізюМбРэТлХфЗНLi6.25PTeS4.25Cl0.75ЁЃTeВПЗжШЁДњSЮЛгажњгкРЉПэяЎРызгЕФРЉЩЂЦПОБЃЌДѓЗљЖШЬсЩ§СђвјерПѓаЭЕчНтжЪЕФяЎРызгЪфдЫФмСІЁЃ

ЭМ3.ЃЈaЃЉLi6PS5Cl,Li6.25PS5.25Cl0.75, Li6.25PTe0.125S5.125Cl0.75ЕФЪвЮТЕчЛЏбЇзшПЙЭМЦзЁЃвдМАБфЮТЕчЛЏбЇзшПЙЭМЦзЁЃ(b) Li6.25PTe0.125S5.125Cl0.75ЕФдкЕЭЮТЛЗОГЯТЕчЛЏбЇзшПЙЭМЦзЁЃ(c) Arrhenius ЙиЯЕlog(ІбT) vs. 1000/TЃЌЪЕбщгыМЦЫуЯрБШНЯЁЃ
дкЧАУцРэТлЩшМЦЕФжИЕМЯТЃЌЮвУЧПЮЬтзщГЩЙІжЦБИаТаЭLi6.25PTeS4.25Cl0.75СђвјерПѓЕчНтжЪЁЃЕБTeВПЗжШЁДњSКѓЃЌШчЭМ3ЃЈaЃЉЫљЪОЃЌРызгЕчЕМТЪЕФгазХУїЯдЬсИпЁЃLi6.25PTeS4.25Cl0.75ЕФЪвЮТРызгЕчЕМТЪИпДя4.5 mS cm-1ЃЌЧвРЉЩЂМЄЛюаЁЃЌНіЮЊ0.160 eVЃЌБЃеЯдкГЌЕЭЮТЛЗОГЯТЃЈ-20ЁцЃЉЃЌяЎРызгЕчЕМТЪШдЮЊ1.612 mS cm-1ЃЌШчЭМ3ЃЈbЃЉЫљЪОЁЃAIMDдЄЙРЕФРызгРЉЩЂМЄЛюФмЮЊ0.168 eVгыЪЕВтЪ§Он0.160 eVЭъШЋздЧЂЁЃШчЃЌЭМ3ЃЈcЃЉЫљЪОЁЃ
ЫЋЗДИЦюбПѓЕчНтжЪЬхЯЕЕФРэТлЩшМЦКЭЪЕбщжЦБИЃК
гЩгкЯжгаЕФСђЛЏЮяЕчНтжЪЯрЖдН№ЪєLiЛђNaИКМЋМфДцдкбЯжиЕФИБЗДгІЃЌЛсБЛЗжНтГЩLi3P, Li2S, Na3P, Na2SЕШдуИтЕФРызгЕМЬхЁЃиНашвЛРргыН№ЪєLiЛђNaИКМЋЕФМцШнадгХСМЕФЕчНтжЪЬхЯЕЁЃвбжЊЕФЕЅаЭЗДИЦюбПѓЕчНтжЪLi3OCl, Li3OBr,Na3OBr, Na4OI2ОпгаНЯКУЕФгыН№ЪєLiЛђNaИКМЋЕФМцШнадЃЌЕЋЪЧЦфРызгЕчЕМТЪНЯВюЁЃвђДЫЛљгкВФСЯЛљвђзщЗНЗЈЃЌдкЕЅЗДИЦюбПѓНсЙЙЕФЛљДЁЩЯЃЌЭЈЙ§НсЙЙИФадЃЌЩшМЦВЂжЦБИаТаЭЫЋЗДИЦюбПѓЕчНтжЪЁЃ
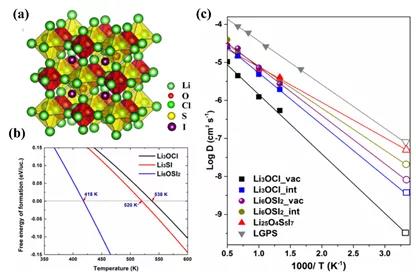
ЭМ4. (a) Li3O0.5S0.5IЕФОЇЬхНсЙЙЭМЁЃ(b) Li3O0.5S0.5IЕФМЊВМЫЙздгЩФмЁЃ(c) ЭЈЙ§AIMDФЃФтМЦЫуИїИіГЩЗжЕФяЎЕФЪфдЫФмСІКЭРЉЩЂМЄЛюФмЁЃ
ЫЋаЭЗДИЦюбПѓLi6OSI2ЕФОЇЬхНсЙЙЃЌШчЭМ4(a)ЫљЪОЃЌбѕСђЙВеМбѕЮЛЃЌаЮГЩЫЋаЭЗДИЦюбПѓLi6OSI2НсЙЙЁЃЭМ4 (b)ЭЈЙ§МЊВМЫЙздгЩФмМЦЫуLi6OSI2зЊБфЮЊЮШЖЈЯрЕФЮТЖШзЊБфЕуЮЊ418KЁЃЭМ4 (c)ЭЈЙ§AIMDМЦЫуЕФБцЮіЯргІЕчНтжЪяЎРызгЕФЪфдЫФмСІ,УїШЗИЛяЎЯрОпБИИќгХЕФРызгЪфдЫФмСІЁЃ
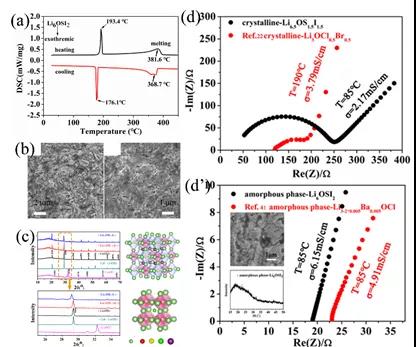
ЭМ5. (a) ЫЋаЭЗДИЦюбПѓLi6OSI2ЕФDSCЮќЗХШШЧњЯпЁЃ(b)жЦБИЬеДЩЬхБэУцКЭКсЖЯУцЕФЩЈУшЭМЦЌЁЃ(c)ЪЕбщXRDЭМЦзгыМЦЫуXRDЭМЦзЁЃ(d)ОЇЬхLi6.5OS1.5I1.5ЕФзшПЙЭМЦзЁЃ(dЁЏ)ЭЈЙ§ЗЧОЇЛЏЯћГ§ПХСЃНчУцКѓЕФзшПЙЭМЁЃ
ИљОнDSCЗжЮіЃЌЭМ5 (a)ЃЌLi6OSI2зЊБфЮЊЮШЖЈЯрЕФЮТЖШдк176.1ЁцЃЌгыРэТлдЄЙР418 K ЃЈ145 ЁцЃЉЛљБОвЛжТЁЃЭЈЙ§ЩеНсШШДІРэЃЌЕУЕНLi6OSI2ОЇЬх, ЭМ5 (c)ЁЃЭЈЙ§ЗЧОЇЛЏКѓЭМ5 (dЁЏ),УїЯдМѕШѕОЇНч/ОЇСЃзшПЙЕФгАЯьЃЌРызгЪфдЫФмСІУїЯдгХгкЕЅаЭЗДИЦЬЌПѓЕчНтжЪЁЃ

ЭМ6. (a)Na3O0.5S0.5IЫЋЗДИЦюбПѓНсЙЙ(b) Na3O0.5S0.5IЕФЮШЖЈЕчбЙДАПк(c)ЗжзгЖЏСІбЇNa+РызгЪфдЫЙьМЃЁЃ
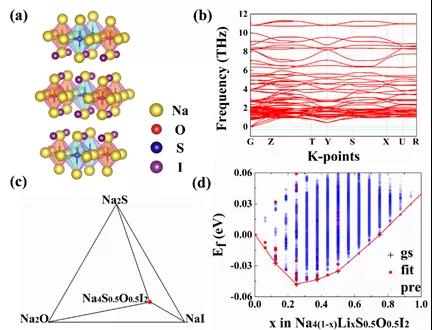
ЭМ7.ЃЈaЃЉNa4S0.5O0.5I2ВузДЗДИЦюбПѓНсЙЙЁЃЃЈbЃЉNa4S0.5O0.5I2ЕФЩљзгЦзЁЃЃЈcЃЉNa4S0.5O0.5I2ЕФЦНКтЯрЭМЁЃЃЈdЃЉЭЈЙ§ATATЭХДиеЙПЊЃЌЫбЫїNa4(1-x)LixS0.5O0.5I2ГЩЗжЯрЭМЁЃ
ЛљгкВФСЯЛљвђзщИпЭЈСПЫуЗЈЃЌРэТлЩшМЦГіNa3O0.5S0.5IЫЋаЭЗДИЦюбПѓ(ЭМ6)КЭNa4S0.5O0.5I2ВузДЗДИЦюбПѓ(ЭМ7)СНжжФЦРызгЕчНтжЪЬхЯЕЁЃетСНРрГЩЗжОљФмЭЈЙ§ФмСПЮШЖЈадКЭЖЏСІбЇЮШЖЈадЕФБцЮіЁЃадФмЗНУцЃЌгыФЦЛЏбЇаджЪЮШЖЈЃЌОпБИгХСМЕФФЦРызгЪфдЫФмСІЁЃШчЭМ8ЫљЪОЃЌвдВузДЗДИЦюбПѓNa4S0.5O0.5I2ЙЙжўвЛЬхЛЏШЋЙЬЬЌФЦРызгЕчГиВпТдЃЌМШПЩвдГЩЮЊвЛИіздЮвМцШнЕФВФСЯЯЕЭГЃЌгжПЩзїЮЊМЋКУЕФЕчНтжЪКЭИКМЋЃЌПЩФцФмСПУмЖШГЌЙ§320 wkg-1ЁЃЯрЙиЪЕбще§дкПЊеЙжаЁЃ
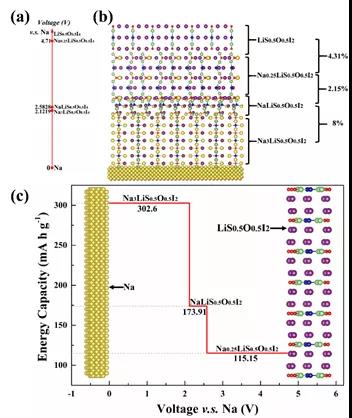
ЭМ8. вдNa4S0.5O0.5I2ЮЊЛљДЁЃЌЙЙжўвЛЬхЛЏЙЬЬхЕчГиЯЕЭГЁЃ
ЛљгкСђвјерПѓаЭLi6PA5XЕчНтжЪЕФШЋЙЬЬЌЕчГивЛЬхЛЏВпТдЃК
ЕчНтжЪгыЕчМЋЕФМцШнадЃК
СђЛЏЮяЕчНтжЪЬхЯЕЃЌЦфгХвьЕФяЎРызгЕчЕМТЪКЭбгеЙадЪЙЦфдкЙЬЬхЕчГижаОпгаЙуРЋЕФгІгУЧАОАЁЃЕЋЪЧгЩгкСђЛЏЮяЙЬЬЌЕчНтжЪЕФЕчЛЏбЇДАПкеЃЌгыИпЕчбЙе§МЋВФСЯжБНгНгДЅЪБЃЌШнвздкНчУцДІаЮГЩяЎРызгЕФКФОЁВуЁЃВЂЧвЃЌСђЛЏЮяЕчНтжЪгыLiН№ЪєИКМЋМфДцдкНЯЮЊбЯжиЕФИБЗДгІЁЃМЋДѓЕФзшАСЫСђЛЏЮяЕчНтжЪдкШЋЙЬЬЌЕчГижаЕФдЫгУЁЃ
ЭМ9.ЃЈaЃЉЭЈЙ§AIMDМЦЫуЃЌБэУїLi6PS5ClгыН№ЪєLiИКМЋЕФИБЗДгІЁЃЃЈbЃЉ ЛљгкЦНКтЯр(0K)ЕФDFTМЦЫуВЛЭЌЛЏКЯЮяЬхЯЕЕФЕчбЙДАПкЁЃ(Y. Zhu, X. He, Y. Mo, A. C. S.Appl, Mater. Interfaces 2015, 7, 23685)ЃЈcЃЉРћгУATATЭХДиеЙПЊМЦЫуSSEдкЧЖяЎгыЭбяЎЙ§ГЬжаЫљаЮГЩЕФВЛЭЌНсЙЙЁЃ
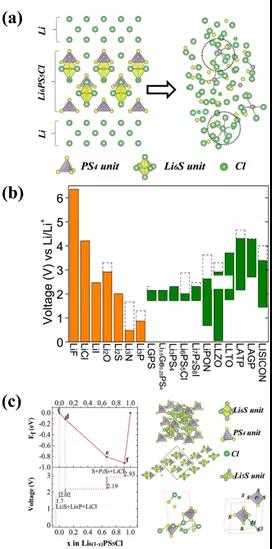
ШчЭМ9 (a)ЫљЪОЃЌЮвУЧЭЈЙ§ЙЙдьLi6PS5Cl|LiЕФвьжЪФЃаЭЃЌЗЂЯжЮЊLi6PS5ClгыН№ЪєLiИКМЋМфЛсЗЂЩњбЯжиИБЗДгІЃЌЕчНтжЪЛсБЛЗжНтГЩLi3PЕШжаМфВњЮяЁЃLi6PS5ClЕчНтжЪЛЙдЕчЮЛНЯИпЃЌдМЮЊ 1.7 V(ШчЭМ9 (b)ЫљЪО)ЁЃЙЪЦфЕчЛЏбЇЕчЮЛгыLiВЛМцШнЁЃСэвЛЗНУцЃЌЕБЕчНтжЪДІгкИпбЙЧщПіЪБЃЌLi6PS5ClЛсбЯжиЪЇШЅяЎЃЌЩњВњРрЫЦP2S5,LiCl,SЕШЪфЕМяЎРызгФмСІМЋВюЕФжаМфЯрВњЮяЃЌ(ШчЭМ9 (c)ЫљЪО)ЁЃ
вђДЫЃЌЮвУЧгаеыЖдадЕФЩшМЦСЫLi6PO5ClКЭLi6PO4SClСНжжЙІФмадЛКГхВуВФСЯЃЌ(ШчЭМ10 (a)ЫљЪО)ЁЃЭЈЙ§МЦЫуЗЂЯжв§ШыВПЗжOКѓЃЌЖдяЎЕФЮШЖЈадДѓЗљЖШЬсЩ§ЃЌШчЭМ10 (b)ЫљЪОЁЃЖјЧвЛКГхВуВФСЯгыЕчНтжЪЬхЯЕОпБИНќЫЦЕФЛЏбЇзщГЩЃЌгажњгкБмУтЛКГхВуВФСЯгыЕчНтжЪМфЗЂЩњбЯжиЕФЛЏбЇИБЗДгІЁЃЖјЧвЃЌЛКГхВуВФСЯгыЕчНтжЪМфОпгаЯрНќЕФОЇИёГЃЪ§ЃЌШчLi6PO4SCl/Li6PS5Cl/Li6PO5ClОЇИёЪЇХфЖШНіЮЊ1.5%КЭ3.1%ЃЌСМКУЕФОЇИёЦЅХфЙиЯЕБЃеЯНчУцДІФкгІСІаЁЁЃгажњгквдLi0.25MnO2|Li6PO5Cl|Li6PO4SCl|LiЕФаЮЪНЙЙжўвЛЬхЛЏШЋЙЬЬЌЕчГиЁЃ
ЭМ10.ЃЈaЃЉЭЈЙ§ATATЫбЫїO/SЙВВєЫљЕУЕНЕФИДКЯГЩЗжЁЃЃЈbЃЉ) зѓСаЮЊLi6PO5Cl|LiЁЂLi6PO4SCl|LiКЭLi6PO5Cl|xLi6PO4SClЕФвьжЪФЃаЭЁЃгвСаЮЊОЙ§AIMDдкИпЮТГЄЪБМфбнЛЏКѓЕФвьжЪФЃаЭЁЃЃЈcЃЉЩшМЦИпФмСПУмЖШЕФвЛЬхЛЏШЋЙЬЬЌЕчГиЁЃ
ЁОБОЮФзмНсЁП
вдВФСЯЛљвђзщЗНЗЈЮЊжИЕМЃЌЮвУЧВЩгУDFTИпЭЈСПМЦЫуЃЌеыЖдВФСЯФмСПЮШЖЈадЃЌНсЙЙЮШЖЈадзіГіЙиМќдЄХаЃЌжИЕМЪЕбщКЯГЩЁЃВЂЖдРызгЕчЕМТЪЁЂРЉЩЂМЄЛАФмЁЂбѕЛЏЛЙдЕчЮЛЕШЕчНтжЪЛђНчУцЙиМќЮЪЬтзіГіШЋУцЕФПЦбЇдЄЙРЁЃРэТлгыЪЕбщЯрНсКЯЃЌМгЫйвЛЬхЛЏШЋЙЬЬЌЕчГиЕФЪЕгУЛЏНХВНЁЃ
ЮФЯзСДНгЃК
https://onlinelibrary.wiley.com/doi/full/10.1002/eem2.12053
ЁОЭиеЙЁП
ИУТлЮФЕФдкжЃжнДѓбЇЙњМвМЖЬиЦИНЬЪкЩлЙњЪЄжИЕМЯТЭъГЩЃЈЭЈбЖзїепЃЉЃЛжЃжнДѓбЇЃЌаьКьНмЃЈВЉЪПбаОПЩњЃЉЃЌЮЊЕквЛзїепЃЛжЃжнДѓбЇЃЌгкгёШЛЃЈЫЖЪПбаОПЩњЃЉЃЌЮЊЙВЭЌЕквЛзїепЃЛжЃжнДѓбЇЃЌЭѕзПЃЈИБНЬЪкЃЉЙВЭЌЭЈбЖзїепЁЃИУЙЄзїдкздШЛПЦбЇЛљН№ЮЏЁЂжЃжнДѓбЇВФСЯбЇдКЁЂжЃжнаТЪРМЭВФСЯЛљвђзщЙЄГЬбаОПдКЃЈhttp://www.zmgi.net/ЃЉзЪжњЯТЭъГЩЁЃИааЛEEMЖдРэТлЬНЫїЙЄзїЕФжиЪгЁЃЭЌЪБЃЌвЊИааЛжЃжнЁЂмўбєеўИЎЖдВФСЯЛљвђдКЕФДѓСІжЇГжЃЌЪЙЮвУЧФмЙЛЭЈЙ§дДЭЗДДаТЁЃИааЛЮхжлИпадФмМЦЫуЃЌЖдБОПЮЬтЬсЙЉЕФЗўЮёЦїММЪѕЗНУцЕФжЇГжЁЃ
ЩлЙњЪЄЃЌЙњМвМЖЬиЦИНЬЪкЁЃдјШЮжАгЂЙњШјРћДѓбЇзЪЩюбаОПЙйЃЛВМТГФЮЖћДѓбЇВФСЯбЇИБНЬЪкЃЛгЂЙњВЉЖћЖйДѓбЇМЦЫуВФСЯбЇНЬЪкЁЂаТФмдДбаОПЫљЫљГЄЁЂЙЄГЬдКдКГЄЁЂРэЙЄМАЬхг§бЇВПжїШЮЕШЃЛгЂЙњВФСЯЛЏбЇЮЏдБЛсЮЏдБЁЂПЩГжајФмдДВФСЯЙЄзїзщГЩдБЁЃ2010ФъШыбЁЙњМвЁАЧЇШЫМЦЛЎЁБЃЌЗўЮёгкжЃжнДѓбЇЃЌДДНЈСЫжагЂФЩУзЖрЙІФмВФСЯбаОПжааФЃЈ2012ЃЉЃЌВЂБЛШЯЖЈЮЊКгФЯЪЁЕЭЬММАЛЗОГВФСЯЙњМЪСЊКЯЪЕбщЪвЃЈ2014ЃЌ ПЦММЬќЃЉЁЂ ЙњМвМЖЕЭЬММАЛЗБЃВФСЯжЧФмЩшМЦЙњМЪСЊКЯЪЕбщЪвЃЈ2015ЃЌПЦММВПЃЉЁЃ2016ФъгкЁАжаджЧЙШЁБДДНЈжЃжнаТЪРМЭВФСЯЛљвђзщЙЄГЬбаОПдКЁЃДДАьСЫЙњМЪЦкПЏЁЖEnergy & Environmental Materials (EEM)ЁЗЃЌгЩJohn Wiley & Sons, IncГіАцЁЃбаОПМЏжагкЖрГпЖШВФСЯФЃФтМАжЧФмВФСЯЩшМЦЁЂФЩУзМАБЁФЄВФСЯжЦБИММЪѕЁЂЯШНјВФСЯБэеїЁЂаТФмдДМАЛЗОГЧхНрВФСЯММЪѕЕШЁЃЗЂБэАќРЈЁЖNatureЁЗдкФкЕФЙњМЪжјУћЦкПЏТлЮФ200грЦЊЃЌЩъЧыВЂЛёЕУЙњФкЭтзЈРћЖрЯюЁЃ
аХЯЂРДдДЃК
- ЩЯвЛПюЃК ЗЂеЙНЯПьЕФЖўЮЌВФСЯЁЊЁЊЁЖChinese Ch
- ЯТвЛПюЃК Bioactive Materials |