
【Research Background】
Alkali metal ion batteries are widely used in portable electronic devices and electric vehicles. The current bottleneck restricting the application of alkali metal ion batteries is potential safety hazards, which are closely related to the flammable and explosive liquid organic electrolytes used in traditional batteries. . The development of all-solid-state batteries based on inorganic solid electrolytes (SSE) is an effective way to solve potential battery safety issues. At present, solid electrolyte systems are roughly divided into two categories: oxide electrolytes and sulfide electrolytes. Oxide electrolytes have the advantages of better stability, insensitivity to air and moisture, and a wider electrochemical window. But there are also problems such as low ionic conductivity, high grain boundary / grain resistance, easy brittle fracture, and high sintering temperature. The sulfide electrolyte has the characteristics of ultra-high ion conductivity, good ductility, small grain boundary / grain resistance, and low preparation sintering temperature. But its short board is also obvious, for example: narrow voltage window, poor compatibility with high-voltage positive electrode materials, poor stability with metal Li negative electrodes, and sensitivity to air and humidity. Exploring a new type of fast ion conductor with better overall performance has become one of the key issues to be solved urgently. In addition, the stability, compatibility, and matching of the solid-solid interface in the all-solid battery still seriously restrict the electrochemical performance of the all-solid battery.
From the huge database, considering the infinite combination of chemical components, looking for a new type of solid electrolyte system, it is difficult to quickly and accurately find electrolyte formulations with excellent performance through trial and error. On the other hand, for the problem of solid-solid interface in all-solid-state batteries, it requires expensive in-situ testing to distinguish clearly. It is difficult to grasp the key scientific issues only by subjective assumptions. Therefore, a set of objective research methods is urgently needed to accelerate the exploration of new electrolytes and provide a rational solution for the construction of integrated all-solid-state batteries. Professor Shao Guosheng‘s research group of Zhengzhou University, through the integration of material genomics methods, has formed a complete set of DFT algorithms for key problems in solid-state batteries, performing high-throughput calculations, exploring new solid electrolytes, and providing solid-solid interface compatibility that solves all solid-state batteries The rationality of the plan provides a strong theoretical support for subsequent experimental research and development.
【Achievement Introduction】
The research team of Professor Shao Guosheng of Zhengzhou University published an article entitled " First Principle Material Genome Approach for All Solid-State Batteries " on Energy Environ. Mater .
【Graphic introduction】
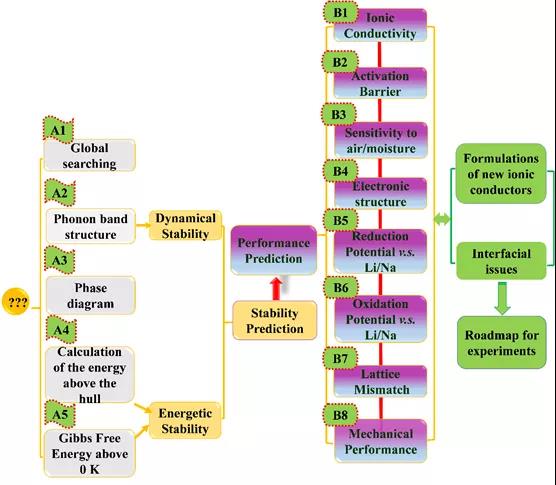
Figure 1. Material genome method flow.
The high-throughput material genomic method calculation process, shown in Figure 1, includes (A) thermodynamic and kinetic stability prediction, (B) solid electrolyte key performance prediction, two parts. The material genomic methods covered in part (A) include: USPEX genetic algorithm, phonon band calculation, phase diagram analysis, 0 K formation energy calculation, including Wen Gibbs free energy calculation. Fully estimate the structural stability and thermodynamic stability of the material to be designed to ensure the feasibility of the designed material in the subsequent work. Part (B), objective prediction of key properties including ionic conductivity, diffusion activation energy, electronic structure, redox potential, lattice mismatch, mechanical properties, etc. The solid-solid interface problem, improve the rationalization plan and theoretical support.
Theoretical design and experimental preparation of the sulfur-silver-germanium-type Li 6 PA 5 X electrolyte:

Figure 2. (a) Li + ion diffusion channel and transport bottleneck in Li 6 PS 5 X. (b) Theoretical phase diagram of Li 6 PS 5 Cl . (c) The phase transition temperature is calculated from Gibbs free energy. (D) Predict the structural stability of Li 6 PTeS 4 Cl through phonon band calculation . (e) , (f) Use the CI-NEB calculation method to clarify the correspondence between the modification scheme and lithium ion diffusion. (G) Estimate the lithium ion transport capacity through AIMD calculation.
In the configuration of bisulfite- germanium Li 6 PS 5 X electrolyte, there is a bottleneck of SXS triangle (as shown in Figure 2a ) , which greatly limits the rapid transport of lithium ions. Therefore, it is urgent to modify and break through the bottleneck of ion transport, which is the key to realize the rapid transport capacity of lithium ions. This part of the work provides a reasonable plan for the sulfur-silver-germanium mineral electrolyte . And make a full estimate of its stability and ion transport capacity. The details are as follows: Figure 2 (b), phase diagram analysis, referring to Li 3 PS 4 and LiCl, Li 6 PS 5 Cl is the metastable phase; through Gibbs free energy calculation ( Figure 2 (c) ), it is found that when the temperature is high At 614K, Li 6 PS 5 Cl will transform into the most stable phase, providing a heat treatment scheme for subsequent experimental work. Through phonon band calculation ( Figure 2 (d) ), no imaginary frequency indicates that the structure is stable. If it is unstable, it indicates that the designed structure is unstable and a phase transition will occur. For the key ion transport performance of the electrolyte, ( Figure 2 (e)-(g) ), CINEB and AIMD calculation are combined to identify and select the best modification scheme. By comparing the ion conductivity and diffusion activation energy, the best theoretical formula L i6.25 PTeS 4.25 Cl 0.75 was selected. The partial replacement of the S site by Te helps widen the diffusion bottleneck of lithium ions and greatly improves the lithium ion transport capacity of the sulfur-germanium-germanium electrolyte.

Figure 3. (a) Room temperature electrochemical impedance spectra of Li 6 PS 5 Cl, Li 6.25 PS 5.25 Cl 0.75 , Li 6.25 PTe 0.125 S 5.125 Cl 0.75 . And variable temperature electrochemical impedance spectroscopy. (b) Electrochemical impedance spectra of Li 6.25 PTe 0.125 S 5.125 Cl 0.75 at low temperature. (c) Arrhenius relation log (ρT) vs. 1000 / T, the experiment is compared with the calculation.
Under the guidance of the previous theoretical design, our research group successfully prepared a new Li 6.25 PTeS 4.25 Cl 0.75 sulfur silver germanium electrolyte. When partially substituted Te S, as FIG. 3 (a), the ionic conductivity has significantly increased. The room temperature ion conductivity of Li 6.25 PTeS 4.25 Cl 0.75 is up to 4.5 mS cm -1 , and the diffusion activation is small, only 0.160 eV, which guarantees that the lithium ion conductivity is still 1.612 mS cm -1 under ultra-low temperature environment (-20 ℃) , as in FIG. 3 (b) shown in FIG . AIMD‘s estimated ion diffusion activation energy is 0.168 eV and the measured data is 0.160 eV, which is completely consistent. As shown in Figure 3 (c).
Theoretical design and experimental preparation of double reverse perovskite electrolyte system:
Since the existing sulfide electrolytes have serious side reactions with respect to metallic Li or Na anodes, they will be decomposed into poor ion conductors such as Li 3 P, Li 2 S, Na 3 P, and Na 2 S. There is an urgent need for an electrolyte system with excellent compatibility with metal Li or Na anodes. Known single-type anti-perovskite electrolytes Li 3 OCl, Li 3 OBr, Na 3 OBr, Na 4 OI 2 have good compatibility with metal Li or Na anodes, but their ion conductivity is poor. Therefore, based on the material genome method, based on the structure of SLR perovskite, through structural modification, design and preparation of a new type of double reverse perovskite electrolyte.

Figure 4. (a) Li 3 O 0.5 S 0.5 I crystal structure diagram. (b) Gibbs free energy of Li 3 O 0.5 S 0.5 I. (c) AIMD simulation to calculate the lithium transport capacity and diffusion activation energy of each component.
The crystal structure of the double-type inverse perovskite Li 6 OSI 2 is shown in FIG. 4 (a). Oxygen and sulfur occupy the oxygen site together to form the double-type inverse perovskite Li 6 OSI 2 structure. Figure 4 (b) The temperature transition point of Li 6 OSI 2 into a stable phase calculated by Gibbs free energy is 418K. Figure 4 (c) AIMD calculations identify the transport capacity of the corresponding electrolyte lithium ions, and it is clear that the lithium-rich phase has better ion transport capacity.

Figure 5. (a) The DSC endothermic and exothermic curve of the dual-type anti-perovskite Li 6 OSI 2 . (b) Prepare scanned pictures of the surface and cross section of the ceramic body. (c) Experimental XRD pattern and calculated XRD pattern. (d) Impedance spectrum of crystal Li 6.5 OS 1.5 I 1.5 . (d ‘) Impedance diagram after eliminating the particle interface by amorphization.
According to DSC analysis, Figure 5 (a), the temperature at which Li 6 OSI 2 transforms into a stable phase is at 176.1 ° C, which is basically consistent with the theoretical estimate of 418 K (145 ° C). Through sintering heat treatment, Li 6 OSI 2 crystals are obtained, as shown in Figure 5 (c). Figure 5 (d ‘) after amorphization significantly reduces the effect of grain boundary / grain impedance, and the ion transport capacity is significantly better than that of single-type anti-calcium mineral electrolyte.

Figure 6. (a) Na 3 O 0.5 S 0.5 I double reverse perovskite structure (b) Na 3 O 0.5 S 0.5 I stable voltage window (c) molecular dynamics Na + ion transport trajectory.

Figure 7. (a) Na 4 S 0.5 O 0.5 I 2 layered reverse perovskite structure. (B) of Na . 4 S 0.5 O 0.5 the I 2 of the phonon spectrum . (C) The equilibrium phase diagram of Na 4 S 0.5 O 0.5 I 2 . (D) Search for the phase diagram of Na 4 (1-x) Li x S 0.5 O 0.5 I 2 through ATAT cluster expansion .
Based on the high-throughput algorithm of the material genome, the theoretical design of Na 3 O 0.5 S 0.5 I double-type anti-perovskite ( Figure 6 ) and Na 4 S 0.5 O 0.5 I 2 layered anti-perovskite ( Figure 7 ) two sodium Ionic electrolyte system. Both types of components can be distinguished by energy stability and kinetic stability. In terms of performance, it is chemically stable with sodium and has excellent sodium ion transport capabilities. As shown in Figure 8, the integrated all-solid sodium ion battery strategy with layered anti-perovskite Na 4 S 0.5 O 0.5 I 2 can not only become a self-compatible material system, but also serve as an excellent electrolyte and anode , The reversible energy density exceeds 320 wkg -1 . Related experiments are in progress.

Figure 8. Building an integrated solid battery system based on Na 4 S 0.5 O 0.5 I 2 .
All-solid battery integration strategy based on sulfur silver germanium-type Li 6 PA 5 X electrolyte:
Compatibility of electrolyte and electrode:
The sulfide electrolyte system, with its excellent lithium ion conductivity and ductility, has broad application prospects in solid batteries. However, due to the narrow electrochemical window of the sulfide solid electrolyte, it is easy to form a depletion layer of lithium ions at the interface when directly contacting the high-voltage positive electrode material. In addition, there are serious side reactions between the sulfide electrolyte and the Li metal anode. Greatly hinders the use of sulfide electrolytes in all-solid-state batteries.

Figure 9. (a) Calculation by AIMD shows the side reaction of Li 6 PS 5 Cl with metal Li anode . (B) Calculate the voltage window of different compound systems based on the DFT of the equilibrium phase (OK). ( Y. Zhu, X. He, Y. Mo, ACSAppl, Mater. Interfaces 2015, 7, 23685 ) (c) Using ATAT cluster expansion to calculate the different structures formed by SSE during lithium insertion and lithium removal.
As shown in Fig. 9 (a), by constructing a heterogeneous model of Li 6 PS 5 Cl | Li, we found that a serious side reaction will occur between Li 6 PS 5 Cl and the metal Li anode, and the electrolyte will be decomposed into Li 3 P And other intermediate products. The reduction potential of Li 6 PS 5 Cl electrolyte is high, about 1.7 V (as shown in Figure 9 (b)). Therefore, its electrochemical potential is not compatible with Li. On the other hand, when the electrolyte is under high pressure, Li 6 PS 5 Cl will seriously lose lithium, producing intermediate phase products like P 2 S 5 , LiCl, S, etc. that have extremely poor lithium ion transport capabilities, (as shown in Figure 9 (c ) Shown).
Therefore, we designed Li 6 PO 5 Cl and Li 6 PO 4 SCl functional buffer layer materials in a targeted manner (as shown in Figure 10 (a)). Through calculation, it is found that the introduction of part O greatly improves the stability of lithium, as shown in Figure 10 (b). Moreover, the buffer layer material and the electrolyte system have similar chemical compositions, which helps to avoid serious chemical side reactions between the buffer layer material and the electrolyte. Moreover, the buffer layer material and the electrolyte have similar lattice constants, such as Li 6 PO 4 SCl / Li 6 PS 5 Cl / Li 6 PO 5 Cl lattice mismatch is only 1.5% and 3.1%, good lattice The matching relationship ensures that the internal stress at the interface is small. It is helpful to build an integrated all-solid battery in the form of Li 0.25 MnO 2 | Li 6 PO 5 Cl | Li 6 PO 4 SCl | Li .

Figure 10. (a) Searching for the composite composition of O / S co-doping by ATAT. (B) ) The left column of Li . 6 PO . 5 CI | of Li, of Li . 6 PO . 4 SCl | of Li and of Li . 6 PO . 5 CI | xLi . 6 PO . 4 hetero model of SCl. The right column is the heterogeneous model after AIMD has evolved at high temperature for a long time. (C) Design an all-solid-state battery with high energy density.
【Summary of this article】
Using material genomics as a guide, we use DFT high-throughput calculations to make key predictions about material energy stability and structural st



