
��ѯ���ߣ�
17715390137
18101240246
18914047343
�ʼ���mxenes@163.com
ɨ���ע�����������ںţ�
������Fronrier
��ע�������½���ϵ���ǣ�
������ҵ�š�
רҵ��������

��10ƪNatureϵ�ж����������ۼ�����δ���������
���ʵĿ��гɹ�������Դ�ڶԿ�ѧ������ʵ�̽������˳���ʵ���о��������о�Ҳ������Ҫ�����ۼ����ܹ���������IJ�������������ʽ���ģ�⣬������ʾ��ѧ������ϵͳ�Լ����Ϸ�����������ǴӲ��Ϻͻ�ѧ����������10ƪ������Nature�����ӿ����漰����ģ��İ������£�����Ҳο���
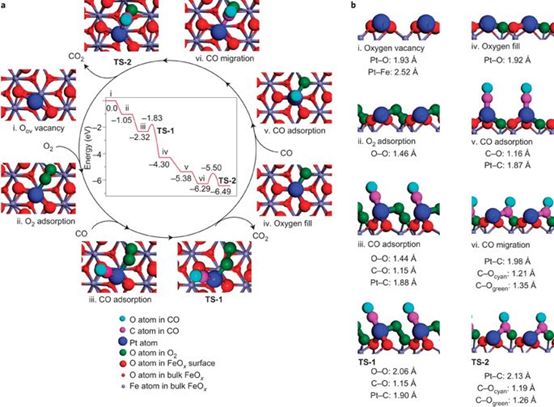
1. ���ο����� Nature Chemistry: FeOx���صĵ�ԭ�Ӳ���Pt����������CO������Ӧ
�п�Ժ������ѧ�����о��������о�ԱЯ���廪��ѧ�������ڿ�����FeOx���صĵ�ԭ�Ӳ���Pt���������о����֣�Pt�ṩ���Ӹ�FeOx���ȶ��˴�����ʹ���CO������ʾ���˺ܺõĻ��ԡ�DFT��������������Pt������CO�������ܺ�CO�ķ�Ӧ���ݡ�
Qiao B, Wang A, Yang X, et al. Single-atomcatalysis of CO oxidation using Pt 1/FeO x[J]. Naturechemistry, 2011, 3(8): 634.
���Ŵ�ѧ��ѧ����ѧԺ֣��ݥ��л���ۡ�֣�Ϸ�Ƚ������п�Ժ������������ѧ���о�Ա����־�����о�Ա�Լ�������Τ˹������ѧHannu Häkkinen���ں�����ͨ�����³������ɨ������������STM���о�Ag374�����Ŵصı������壬����Ƿ���ˮƽ���߷ֱ棬���DFT���ۼ�����ģ��ʶ��ʵ�ֶԱ���������ò�ͽṹ�Լ��Ŵ�ȡ���ʶ��
Zhou Q, Kaappa S, Malola S, et al.Real-space imaging with pattern recognition of a ligand-protected Ag 374nanocluster at sub-molecular resolution[J]. Nature communications, 2018, 9(1):2948.
3. Nature Chem.�����ۼ�����֤�Ͱ���ʵ�顰����������̬
ʵ����ֱ�ӹ۲⻯ѧ��Ӧ�Ĺ���̬���DZ���Ϊ��ѧ����ġ�ʥ������������̬���ݼ��ţ�ʹ��ʵ����������̬����Ķ���ѧ��ü������ѡ�����̬�����������ۼ���������滥����֤�����Ѿ��ɹ����ڽ�ʾһЩ���ͷ�Ӧ��������Ȥ�Ĺ���̬����ѧϸ�ڡ�
�������ݴ�ѧ��������УDaniel Neumark���ڿ����鷢չ���µĻ����������ٶȳ���Ĺ���뷽�����õ��Ĺ�������ķֱ��ʿɴ�1cm-1����ʵ�鼼������������������Ĺ���̬������ȡ�������ϸ����ѧ��Ϣ��Ȼ����ڸ����ͷ�㣨���Ծ�ȷ���ַ�Ӧ���ݡ���Ӧ�ȵ���Ҫ��Ϣ�������Լ13����ṹ�����ö��ײ������ʽ������������Ϸ����������˸���ϵ��ȫά�ľ�ȷ�����档�ٲ��ü�ά���Ӷ���ѧ�������м����ģ�⣬�õ��Ĺ�������߾��ȵ�������ʵ����������ȷ�ؽ���ʵ��۲�Ĺ�������ס�����ʵ��۲�ķ���Ҫ�����ڲ��︴�����Feshbach����
Weichman M L, DeVine J A, Babin M C, et al. Feshbachresonances in the exit channel of the F+CH3OH�� HF+CH3O reaction observed usingtransition-state spectroscopy[J]. Nature chemistry, 2017, 9(10): 950-955.
4. Thomas Bligaard��Jens K. Nørskov������Nature Communications������ѧϰ��DFT�������Խ�����淴Ӧ������
ThomasBligaard��Jens K.Nørskov�����Ժϳ���(CO+H2)�ڴ���Rh(111)����ķ�ӦΪ��������group additivity fingerprints��Ϲ���̬scalingrelations�ͼķ��������������ģ��(surrogate model)����ʹ�ø����ģ��Ԥ��ϳ�����Ӧ����Ҫ�ķ�Ӧ���ٲ�������ѧϰ�����еĴ��������ܶȷ������ۼ���IJ�ȷ����֮�ڡ�
�������û���ѧϰ�ķ������DFT����ȷ���˺ϳ����������������������ŷ�Ӧ·�����Ի���ѧϰ�����ڼ��������Ӧ��������ʵ���Ե�һ����
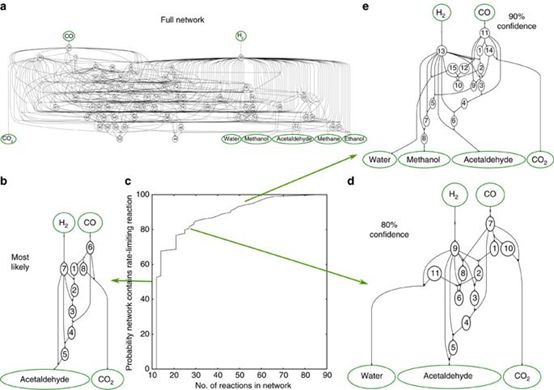
Ulissi Z W, Medford A J, Bligaard T, et al.To address surface reaction network complexity using scaling relations machinelearning and DFT calculations[J]. Nature communications, 2017, 8: 14621.
5. ����ƽ�ȿ����� Nature�� ���������ӿ��Ʋ��ྫȷ��װ����ʯīϩĤ
�й���ѧԺ�Ϻ�Ӧ�������о�������ƽ��JingyeLi���Ϻ���ѧ�������Ŷӡ��Ͼ���ҵ��ѧ�������Ŷӣ���ͬͨѶ������ʹ��K+��Na+��Ca2+��Li+��Mg2+������ʾ�����������ӿ��Ʋ��ྫȷ��װ����ʯīϩĤ�����ֳ����������ɸ�ֺͺ�ˮ�������ܡ����⣬��һ�����������ӿ��Ƶ�Ĥ��������Ч��ѡ���Ե��ų����нϴ�ˮ����������������ӡ�����Ӧ�õ�һ��ԭ��������������չ��ױ��������ȶ�������������λ������������źͷ�������ĵط���
ͨ���ܶȷ������ۼ����������Na+��ȣ�����������Ӧ�þ��б�ʯīϩƬ��ǿ��������-������á�
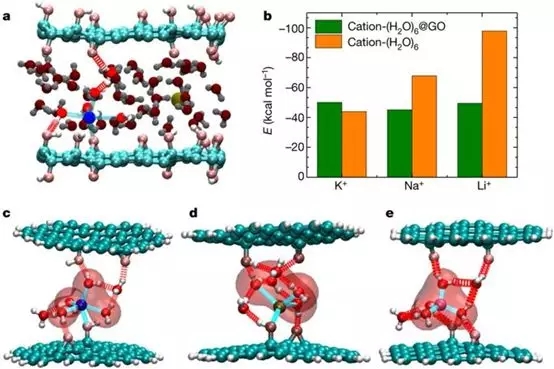
Chen L, Shi G, Shen J, et al. Ion sievingin graphene oxide membranes via cationic control of interlayer spacing[J].Nature, 2017, 550(7676): 380.
6. Nature Materials����Ҫ���õ�C-H�����������ö������ۼ���
˹̹����ѧJens K. Nørskov�����Ŷ���Nature Materials�ϱ�����һ���µķ��������۲�ͬ�������ڻC-H���ϵ��������Ӷ��ṩһ����Ч��ɸѡ������������;��
���û���λ���H����������Ϊһ��ָ�������㲻ͬ�����ı����C-H���Ļ���������߹����˼������������Cu-CHA����ɸ��IrO2/MgO��Co-MOF��Fe-N-C��Au�������ӵȲ�ͬ���͵Ļ���λ���ϵķ�Ӧģ�ͣ��������˷�Ӧ�Ļ�ܺ�EH�����ֹ���̬���ݣ�ETS���ĺ�EH�зdz��õĹ����ԣ��Ӷ�˵����Hydrogen affinity����Ϊһ����ָ�������ͬ��λ��C-H���������ǿ������һ���Ŀɿ��ԡ�
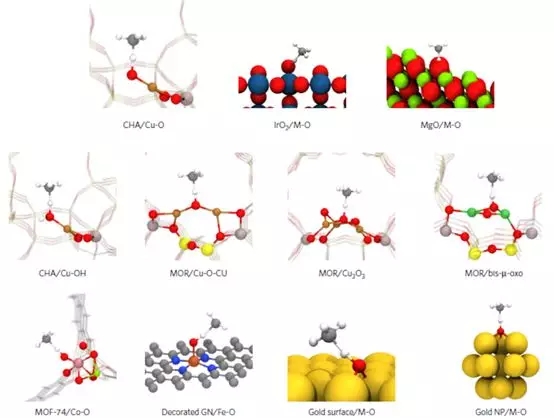
Latimer A A, Kulkarni A R, Aljama H, et al.Understanding trends in C�CH bond activation in heterogeneous catalysis[J].Nature materials, 2017, 16(2): 225.
7. Jianwei Miao ������ Nature���ڵ�ԭ��ˮƽ�Ͻ��ܻ�ѧ����/����̬ͬ�������ʵĹ�ϵ
���������Ǵ�ѧ����ϵͳ�о�����Jianwei Miao�����Ŷ�ȷ����һ����6569����ԭ�Ӻ�16627����ԭ����ɵ���-������������ԭ�ӵ���ά���꣬������ԭ��ˮƽ�Ͼ�ȷ���������Ļ�ѧ����/����̬�;���ȱ�ݶԲ������ʵ�Ӱ�졣
���Ŷӻ������˲��Ϸḻ����ά�ṹϸ�ڣ��������е�ԭ�ӳɷ֡��������硢���ྦྷ�硢��λ��ȱ�ݺͽ���ȱ�ݡ����Ŷ��о�������ͨ��ֱ������DFT�����в��ϵ����ʣ���ԭ������������ž��Լ��ֲ��ž��������ԣ�����ʵ����22Ƥ�뾫���²ⶨ�����е�ԭ������ͻ�ѧ�ɷ֡�
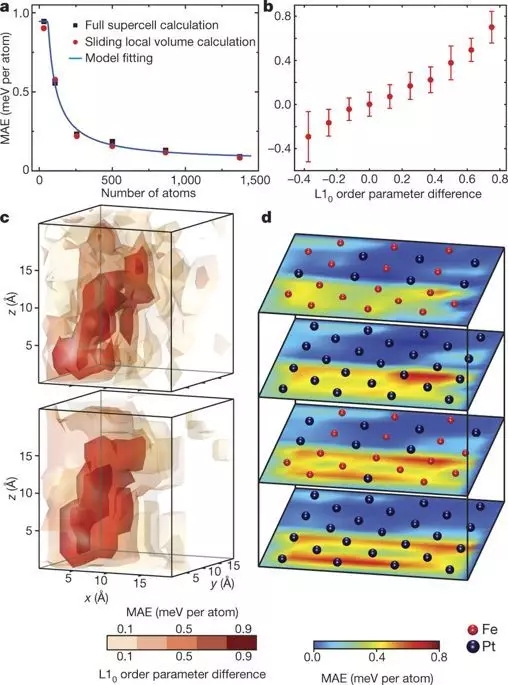
Yang Y, Chen C C, Scott M C, et al.Deciphering chemical order/disorder and material properties at the single-atomlevel[J]. Nature, 2017, 542(7639): 75.
Antonietta Guagliardi �� Norberto Masciocchi�����������µ�X-rayɢ�似���Լ�DFT�о����ڼ�С�߶��£�<8nm��PbS��PbSe���ӵ㾧��Ť���Լ�Pb �Ǿ���ȡ����
��������DFT֤���ˣ�1.����Pb�Ǿ�����[111]�᷽���ȡ�����¾���ṹ�����νṹ��ǶԳƵ�������ϵת�䣻2.�ھ���ṹ����ʱ��Pb-S���е�����������������ǶԳƵ�������ϵ�еĿ�λ�γ��������½���3.������Ӧ��������PbS��������Ť����
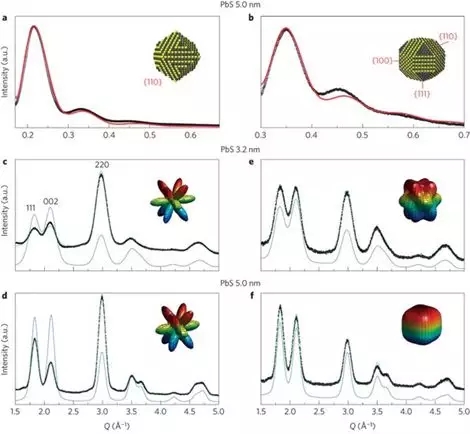
Bertolotti F, Dirin D N, Ib��ñez M, et al.Crystal symmetry breaking and vacancies in colloidal lead chalcogenide quantumdots[J]. Nature materials, 2016, 15(9): 987.
�����ֺ��ܾ��Ŷӷ�����������̼���ϱ��棬ˮ����Ȼ�����ܹ��������ܡ�������Ϊ��һ��ѹ��Ҫ������ˮ���������ƣ�ˮ���������в�����ѹ�����ˮ��������������ˮ���������̿����ˮ�Ӵ�����磨�Ƶ���−33.2 mV����ˮ��������̼���Ͽ��ͷ�϶�����в������ƲDFT�����о���ˮ���ܻ�ʯīϩƬ��Ӵ����ɷֲ�����֤����һ�۵㡣

Xue G, Xu Y, Ding T, et al.Water-evaporation-induced electricity with nanostructured carbon materials[J].Nature nanotechnology, 2017, 12(4): 317.
10. Wonbong Choi�Ŷ�Nature nanotechnology��2D MoS2��Ϊ﮽��������ı�����
Wonbong Choi�Ŷӱ�������﮽����������10 nm���MoS2��Ϊ﮽��������ı����������Ч�����-���ص�ѭ�����ܡ���������DFT����ģ���ڸ�/��Li+Ũ����Li+��MoS2�IJ��Ͳ��е���ɢ����������������������MoS2�������ɢ���ݸ��͡�������ɢ·��������Li+��ɢ�������������֦����
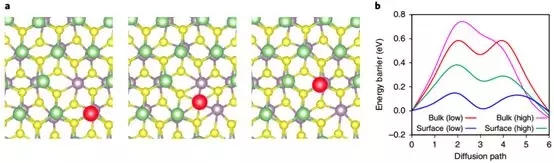
Cha E, Patel M D, Park J, et al. 2D MoS2as an efficient protective layer for lithium metal anodes in high-performanceLi�CS batteries[J]. Nature nanotechnology, 2018, 13(4): 337.
���������ۼ��㻯ѧ�Ŀ��ٷ�չ������ģ���ڲ��ϻ�ѧ�о��е���������㷺�����롣���������Ѿ����γ��ˡ����Ʊ�-����ģ��-�Ƚ����������о�ģʽ������������ʵ��ͼ���ģ���������֤���������������ĵĿɿ��Ժ��Ͻ��ԣ������ܹ��õ����㷺���Ͽɡ�
��ԴSource��
http://www.cailiaoniu.com
http://www.nanoer.net
 |
 |
 |
 |
| ��ά����Frontier | �������ײ���ǰ�� | MXenes Frontier | ����ҽѧFrontier |

| ��ܰ��ʾ�����������²ĿƼ�����Ӧ��Ʒ�����ڿ��У������������塣������վʾ��ͼԴ�Ի�������ͼƬ�����ο�������ʵ�ʲ��Խ��Ϊ��������Ȩ����ϵ��������ɾ������Ʒ���������ο�������ʵ��ֵΪ�� |
|
��Ȩ���� © 2019 ���������²ĿƼ�����˾
All rights reserved. ��ICP��16054715��-2 |
ɨһɨ