
��ѯ���ߣ�
17715390137
18101240246
18914047343
�ʼ���mxenes@163.com
ɨ���ע�����������ںţ�
������Fronrier
��ע�������½���ϵ���ǣ�
������ҵ�š�
רҵ��������

�ܶȷ������������ۻ�ѧ������ȡ�þ�ijɹ����ܹ��ṩ��ϸ�ķ�Ӧ���ڱ��������Ľṹ������λ��������ع�����ϸ��Ϣ��Kohn��Pople���ڸ����������˾������1998���ŵ������ѧ����
DFT����ķ�չʹ�ôӵ���ԭ�ӳ߶�����ʶ�������۽ṹ�뷴Ӧ������Ϊ���ܣ��Ѿ���Ϊ�о���ѧ��ϵ�۽ṹ�ͻ�ѧ��Ӧ��������Ҫ�����ֶΣ����ڸ��õ��������Ӧ���̺ͷ�Ӧ�������Լ���Ƹ�Ч�Ĵ����ṩ�˿ɿ�������֧�֡�
�ڲ��ϼ�������VASP��Ŀǰ���ϼ���ʹ�õ����ձ����������֮һ��������11�������У�ʹ��VASP�������8������ȫ��Ϊ��Vienna Ab-initio Simulation Package������άҲ�ɴ�ѧ��HafnerС�鿪���ġ�VASP�ܽ��е��ӽṹ�ͷ��Ӷ���ѧģ�⣬���Լ�����ϵļ������ʣ����������ǡ��������������ӽṹ���ʣ�����̬�ܶȡ��ܴ��ṹ�������ܶȷֲ������Ӿ�����������ѧ���ʣ���糣�������չ��ס������ʣ��Լ���ѧ���ʵȡ�
VASP����ǿ�̰�֮��������û��һ���ܺõIJ������棬MedeA�ɹ������������⣬����һ�����������Ƚ���������ѧVASP�ͷ��Ӷ���ѧLAMMPS�����һ��ƽ̨����������ʵ����Windowsϵͳ�´���ģ�͡����ò��������ӻ�������������ڲ���ʹ�ã�MedeA����ǿ����û�е�����Դ��
1��Nature Nanotechnology��DFT���㵥ԭ�Ӵ����в�ͬPtԭ�ӵķ�Ӧ·�����о���ԭ�������
�пƴ��о��˹��ڵ�ԭ�Ӵ��еĵ�ԭ������ã��о���Ա����DFT�������㣬��ʾ�˴�CO2�������������Pt��ԭ�Ӽ��Эͬ���á�
����ģ��������������Pt��ԭ��·����CO2�����Ƽ״������в����������м����֣����ٲ���CH2OH*-CH3OH�����CH2OH*Ϊ��Ҫ�м����֡�������Pt��ԭ��·����CO2�����Ƽ״��������м����м����֣����ٲ���COOH*-HCOOH�����COOH*Ϊ��Ҫ�м����֡�
����Pt��ԭ��·�������Pt��ԭ��·���IJ�ͬ���Ӷ���ʾ���ֵ�ԭ���ڴ�����ȫ��ͬ�Ĵ����ܣ���֤����Pt��ԭ��֮���������Эͬ���á�
Li H, Wang L, Dai Y, etal. Synergetic interaction between neighbouring platinum monomers in CO2 hydrogenation[J]. Nature nanotechnology, 2018, 13(5): 411.
2. ʹ��VASP�о�������λ�㣬�����������Ե�����ԭ��
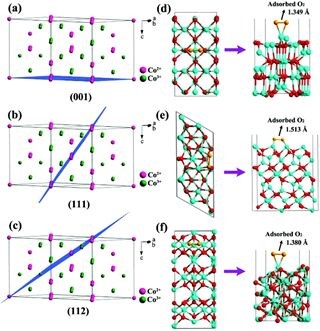
����ѧ�������ı�����Ŷ������һ�ּ�㡢���ٿɿغϳɵ�����ʯīϩ���ز�ͬ�������Co3O4���ṹ���·���������Co3O4����Co2+��Co3+����λ��ı������о��������������Ѹ���Ϊ���ô������ֳ����������ԭ��������˫���ܻ��ԣ�������ǿ����-����������ܡ�
����Ҳ�Dz��ü���ģ�⣨VASP����������ǿ���ܣ������ܶȷ������ۣ�DFT������Co3O4����������϶������Co����λ��ͬʱ��ʾCo3O4�����嵪����ʯīϩ����֮������Эͬ���ã������������Ե�����ԭ��
Han X,He G, He Y, et al. Engineering Catalytic Active Sites on Cobalt Oxide Surfacefor Enhanced Oxygen Electrocatalysis[J]. Advanced Energy Materials, 2018,8(10): 1702222.
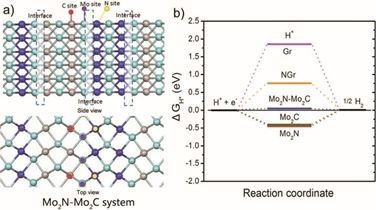
3.����Mo2N-Mo2C��ϵ��ʹ��VASP��������λ���������ȷ������������
��������ѧ����ս��ڣ�ͨѶ���ߣ������Ŷӣ�ͨ��ԭλ�������ԭ����ʯīϩ���صĵ���̼�������ʽ����ڸ�Ч�ĵ�����ⷴӦ��HER����
����ʹ��VASP�����������������ܶȷ������ۣ���������Mo2N-Mo2C��ϵ�Ͽ��ܵ�����λ���Լ����ڸ����о�ϵͳ�����HER�����ܣ�����ָ��Mo2N-Mo2C���洦��N-Mo-C����Ҫ�Ĵ��������ģ����ԭ����ʯīϩ��Ϊ������Ҫ�����Լ���ɴ��ݵ����á�
Yan,Haijing, et al. "Holeyreduced graphene oxide coupled with an Mo2N�CMo2Cheterojunction for efficienthydrogen evolution." Advanced Materials 30.2(2018)
4. ʹ��VASP�����������о���ͬ��ԭ�ӵ�CO��������
���ݴ�ѧ��ѧ����ѧԺ��ɭ������ͨ�����ۼ��㿼���˲�ͬ��ԭ��M( Ag��Au��Pt��Rh��Pd��Fe��Co ��Ir) �����ھ���B��ȱ��h-BN�ϵ�CO���������� ��M-������롢M-N�����ͼ��ܡ�����ת�ơ��������������ܡ�����ӻ��Լ�̬�ܶȣ���˵����ԭ����B��ȱ��h-BN�ļ��ι��ͺ�ǿ����á�
���ۼ���������M-h-BN�Ľ���λ( Fe��Co��Ir ��Pd) ��O2��������CO���ȶ�����CO������Ӧ����E-R����������Co-h-BN �ڵ���CO������Ӧ��������͡�����̬�ܶȷ���������3d�����O2��2p�������ǿ�ӻ���ʹO2������������͡��������������λ ( Cu��Ag��Au��Pt ��Rh) ��CO������ǿ��O2���Ӷ������´���CO�ж���
Lin S, Ye X, Johnson RS, et al. First-principles investigations of metal (Cu, Ag, Au, Pt, Rh, Pd, Fe,Co, and Ir) doped hexagonal boron nitride nanosheets: stability and catalysisof CO oxidation[J]. The Journal of Physical Chemistry C, 2013, 117(33): 17319-17326.
5. ʹ��VASP�о���ԭ�ӱ���������ȷ�����ȶ��ṹ
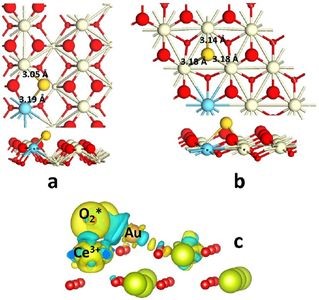
�������������ѧHensen�Ŷ�ʹ��VASP����������DFT����ģ�⣬������Au��CeO2 (110) ���������AuǶ��CeO2 ( 111) �������λ������ǰAu������CeO2�����γɵĶԳƽṹ�Ľ��۲�ͬ�����ָ��ȶ��Ľṹ��Oԭ����Au����ƫ�ƣ���Ce��Au�γ��ż������ֲ��Գƽṹ���������ȶ���
AuǶ��CeO2 ( 111) �������λ������Au-CeO2�ĵ������ţ����Ӵ�Ce��ת�Ƶ�Au��6s������γ�Ce��Au���ۼ��������о���Ա�����֣�CO����������Au������̬������أ�Au���и���̬��CO������ǿ��
Song W, Hensen E J M.Structure sensitivity in CO oxidation by a single Au atom supported onceria[J]. The Journal of Physical Chemistry C, 2013, 117(15): 7721-7726.
6. ����VAS������ʹ��DFT����������ԭ�Ӵ��ķ�Ӧ����
������������ѧ���з��Ŷ��ܵ�ԭ�Ӵ���Pt1 /FeOx�������������˲�ͬ��ԭ��(Au��Rh��Pd��Co��Cu��Ru �� Ti) �ֱ��ص�������λ��������λ��FeOx�����϶�CO������Ӧ��Ӱ�졣
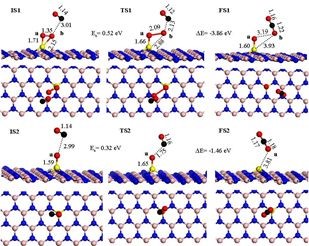
��ԭ�ӵĴ����ܹ���������λ���γ����ݣ��������εȵó�������һ�¡�������λM1/FeOx ( M = Rh��Pd��Co ��Cu) CO������Ӧ���L-H������DFT����������ͼ����Ӧ��Ϊ������
��һ��CO2���γ�: OC* + O-O* ��CO2 + O*��
�ڶ���������CO�뻯ѧ������O��Ӧ�����м����OCO* : CO* + O* ��OCO* ��
�������м����ת���CO2: OCO* ��CO2��
�Բ�ͬ����ȡ�����о���ʾ������ѧ������ѧ�������Rh1/FeOx��Pd1/FeOx������Pt1/FeOx�и��õ�CO���������ܡ�
Ϊ��̽�ִ�����CO ������ͬ�����ܣ����Ƿ��������ĵ��ӽṹ���ԱȲ�ͬMԭ����Χ��ͶӰ̬�ܶȣ�����M1 /FeOx(M = Pt��Rh��Pd �� Ru) ��d��������������ܷ����ƶ�����˴������и���Ŀ�d����Ӷ�CO��O2�������Ӿ��и�ǿ������á������ֿ�����O2������������O2-p �����M-d( M = Pt��Rh ��Pd) ������������ӻ���ʹO2�������CO������
Li F, Li Y, Zeng X C, etal. Exploration of high-performance single-atom catalysts on support M1/FeO xfor CO oxidation via computational study[J]. ACS Catalysis, 2014, 5(2):544-552.
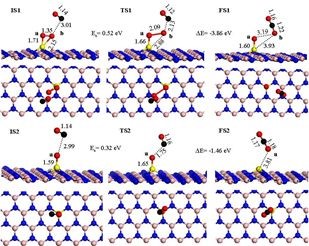
7. ʵ��+DFTģ��̽��Pt /��-Al2O3��CO������Ӧ����
�������������ʵ����Narula�Ŷ��Ʊ��ĵ�ԭ�Ӵ���Pt /��-Al2O3��CO������ͨ��DRIFTS����CO��������ʹ��VASP����������DFT����ģ�⡣
����CO ��O2���������ڵ�ԭ��Pt����λ�����Դ�ͳ��L-H ���������á����ǵ��л�����������о�����͵�ԭ��Pt /��-Al2O3(010)��d10�������( Ph3P)2PtΪ�ȵ����壬������Ż����L-H��������Ӧ��������ͼ��ʾ��
����ͨ��DRIFTS����CO����������ͬPt������������Ʒ���������γ���̼�������м����Ӷ�֤ʵ��CO���������Ż����L-H ������DFT�о���ʾ������������Ӧ�����ϸ��Ե�L-H ��Ӧ��������CO(��NO) ��O2��ͬ������Pt ��ԭ�ӻ������ϲ������巢������; �������۽����ʾPt /��-Al2O3�����ַ�Ӧ�о��������õĴ����ԡ�
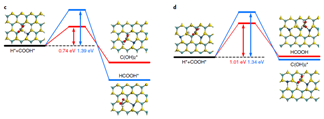
8. ����VASP���������г��DFTģ����ϳ�Pd-Cu˫�����Ͻ��CO2�����Ƽ״�
����������ѧ��С�����ڲ����ܶȷ�������(DFT)������ʵ���о��ķ�������Pd-Cu˫�����Ͻ��CO2����ϳɼ״��ķ�Ӧ���������������о�����ʾ��˫�����Ͻ�ṹ��ˮ��CO2ת�����״�ѡ���Ե���ҪӰ�졣
DFT�����о����������������PdCu(111)������е���λ��Pdԭ�ӱ�¶�ڱ����ϣ����CO2��H2��������ȱ��渻��Cu��ƽ����PdCu3(111)�Ͻ���и��ߵĻ��ԣ�ͬʱ���CO2�ij�ʼ����ת��Ҳ���ֳ�������Ĵ����ܡ�DFT����Ԥ��Pd-Cu�ͺϽ���CO2�����Ƽ״�������������
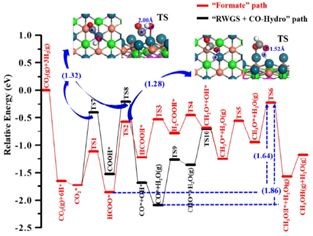
ͨ�����㷴Ӧ·���ͻ�Ԫ��Ӧ����ѧ�������PdCu(111)�����ϼ״����ɵ����Ʒ�Ӧ·����
CO2*��HCOO*��HCOOH*��H2COOH*��CH2O*��CH3O*��CH3OH*
����DFT��������������Ϧ������������ѧ��Դ�о����Ľ��첩ʿ�������ϵͳ��ʵ���о����ϳ��˸���PdCu(111)�Ͻ��༰����PdCu3(111)�Ͻ����˫���������������ۼ���ģ����һ�¡�
Nie X, Jiang X, Wang H, et al.Mechanistic Understanding of Alloy Effect and Water Promotion for Pd-CuBimetallic Catalysts in CO2 Hydrogenation to Methanol[J]. ACS Catalysis, 2018,8(6): 4873-4892.
9. ����VASP��Ϲ���̬������AIMD�����ɹ�����ʵ������ʾ����ѧ�ڶ������Ӧ�е���Ҫ��
���ݴ�ѧ��ѧ����ѧԺ��ɭ��������������DFT����������SAC����λ������ʺ�1,3-����ϩ���ⷴӦ���������ڵڶ���������У������̬����ѧ����Ŀ�����1-��ϩ���Ѹ��������ǽ�һ���������ɶ��飬�Ӷ��ɹ��ؽ�����ʵ��۲쵽�ĸ�ѡ��������
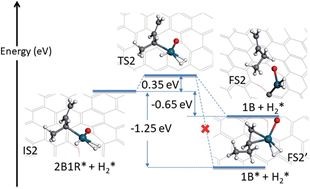
��һ��ӱ�Ĺ۵��ڶ������������ʮ�ֺ����ģ���ʾ�˶���ѧ�ڶ������Ӧ�е���Ҫ�ԡ�����ѡ���Ի���Ҳ����������������ԭ�Ӵ���Ӧ���̣���Ϊ��ԭ���������Ӧ�ṩ��Ч�����ԭ����
Feng Y, Zhou L, Wan Q,et al. Selective Hydrogenation of 1, 3-Butadiene Catalyzed by A Single Pd AtomAnchored on Graphene: The Importance of Dynamics[J]. Chemical Science, 2018.
10. DFT������͵���d������λ���������Co4N��HER���������ȶ��ԡ������ܡ�DOS��
�й���ѧ������ѧ���������ں����������о�Աͨ�����ɽ������ӵ�����Co4N��d������λ�ã��ɹ������������HER�����ԡ�V���ӵ�Co4N����Ƭ��V-Co4N NS����10 mA/cm2�¼��Խ����еĹ����ƿɴ�37 mV��ҪԶ����Co4N�������ӽ��ڻ�Pt/C������
��ʵ��Ļ����ϣ��о���Ա������DFTģ������һ����ʾV���Ӷ�Co4NHER���Ե�����Ӱ�졣
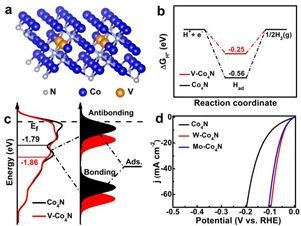
��ϵ�������������ۼ������V�����ܹ�����Co4N��ϵ���������������ѧ���������ģ���ͼa��ʾ��
�����ܼ�����ͼb��ʾ��������������ͼ������V-Co4N������H����GH*��Ϊ-0.25 eV����Co4N��-0.56 eV�����ӽ�����ѧ����ֵ����˵��V�����ܹ��ٽ�����������/�Ѹ����̡�
DOS���㣺�����ܼ�������DOS�仯�������ڱ��������������֮��ĵ�������á��������̬��������ɽ���d̬����֮���������õ����γ��˷���ijɼ�̬�ͷ���̬�����гɼ�̬Զ�ͷ����ܼ�����������ȫ��������������̬���ӵ����ȡ��������ڷ����ܼ�����̬�ͼ���ǿ�ȡ���ͼ4c��ʾ��Co4N��V-Co4N��d�����ģ�Ed��Ϊ-1.79 eV��-1.85eV�������V���Ӻ�d������������ķ�������ƫ�ƣ�����̬�����������������������֮�������ã��ý����UPS���Խ����һ�¡�
���ʵ�������о���Ա֤��ͨ������d������λ�ÿ�����Ч����Co4N��HER�����ԡ�
Chen Z, Song Y, Cai J,et al. Tailoring the d�\Band Centers Enables Co4N Nanosheets To BeHighly Active for Hydrogen Evolution Catalysis[J]. Angewandte ChemieInternational Edition, 2018, 57(18): 5076-5080.
11. ��CP2K���ϴ���OER/ORR���۷����е�Ѥ�����������ܡ�����λ�㡢�����ܣ�CP2K
CP2K�����ӻ�ѧ�������������VASP֮��ļ���ģ���������㡣��������������ѧ���¿�����һ����ѿ�Դ������֧���������뾭�鷽����HF��MP2��DFT��QM/MM�ȶ��ַ������ܽ��е��ӽṹ��CPMD���ṹ�Ż���NEB�ȶ������͵ļ��㡣��CP2K���ϴ�������ײ�������Ļ���
����ʦ����ѧ���������ڡ������������ڣ���ͬͨѶ���ߣ��Ŷӳ������MOF���ϵĽṹ���ƣ���MOF�����ṹ��Ϊ��Ӧ������MOFǰ��������Ʊ�����ò���������������OER/ORR˫���ܵ������
�Ŷ���CP2K�������е�DFT������Co9S8@CT��OER/ORR��Ӧ�����е������ܼ������ܱ仯�����˼���ģ�⣬���ʵ������������������Co9S8@CT��OER/ORR�ķ�Ӧ��������ָ����Co9S8@CT�����������Ե���Դ��
Ϊ�������ھ�Co9S8@CT-800��ORR��OER�ķ�Ӧ�������о���Ա��CP2K�������µ�DFT������Co9S8�������ڵ�����̼����ȱ��λ��������ܡ��������������ڴ�����λ����������Լ���ͬ��λ��OER/ORR��Ӧ�е������ܱ仯�����˼���ģ�⡣
�����ܣ�DFT�����������Co9S8������̼���ܱ����Nԭ�ӿ�λ��C-Nԭ�ӿ�λ��Cԭ�ӿ�λʱ��������ϵ�������ֱ�����1.46 eV��2.38 eV��3.36eV����Co9S8��Ҫ�����ڵ�����̼���ܵ�Nԭ�ӿ�λ�ϡ�
����λ�㣺Ϊ��ȷ��Co9S8@CT-800�����еĻ���λ�㣬�о���Ա�Ժ����������֣�OOH*��O*��OH*����Coλ���Sλ���ϵ����������ܽ����˼���ģ�⣬�������Sλ�㲻�ܺܺõ����������������֡��������������֣�OOH*��O*��OH*���ڵ�����̼�����ϵ������ܼ����������������������Cλ���Nλ���ϵ�����Ҳ���ȶ������Co9S8@CT-800�����Ļ���λ����Ҫ��Coԭ���ṩ��
�����ܣ��ڲ�ͬ��λ�£�Co9S8@CT-800��OER/ORR�������ܱ仯���ᷢ�������ı仯��
��U=0 Vʱ��������̼���ܱ���Nԭ�ӿ�λ�ϵ�Co9S8��pHΪ13�ļ��Ե��Һ�д�ORR�Ĺ�����һ�����ȷ�Ӧ����U=1.23 Vʱ���γ�OOH*��Ҫ�˷�0.31eV�Ļ�ܡ���U=0.79 Vʱ���������ݾ���ʧ�ˣ���ʱORR�е����л�Ԫ��Ӧ������0.44 V�Ĺ���λ���Է����У����Pt��������Ҫ�Ĺ���λ��0.45 V�����١����ֱ���Co9S8@CT-800�ڼ�����Һ�е�ORR������������Pt����������
����Co9S8@CT-800�ڼ��������´���OER����U=1.23 Vʱ�������м���OOH*���γɲ�����Ҫ�˷����Ļ�ܡ�����U=1.62 Vʱ��OER�����еĻ�Ԫ��Ӧ����������·�Ӧ����ʱ��Ӧ�Ĺ���λΪ0.39 V����Щ��������������ʵ�����Ǻϡ���֮��������OER����ORR��OOH*���γɶ���������Ӧ���ٿز��衣Ҫ��һ���������д�����OER/ORR��˫���ܵ�����ԣ�����Ҫ��OOH*���γɲ������֣��Ż�����ֶԺ����������ֵ������ܣ��ڸ��ϴ����IJ�ͬ���֮���γ�Ч�ʸ��ߵ�ЭͬЧӦ��

Liu T, Zhang L, Tian Y.Earthworm-like N, S-Doped carbon tube-encapsulated Co9S8nanocomposites derived from nanoscaled metal�Corganic frameworks for highlyefficient bifunctional oxygen catalysis[J]. Journal of Materials Chemistry A,2018, 6(14): 5935-5943. MLA
 |
 |
 |
 |
| ��ά����Frontier | �������ײ���ǰ�� | MXenes Frontier | ����ҽѧFrontier |

| ��ܰ��ʾ�����������²ĿƼ�����Ӧ��Ʒ�����ڿ��У������������塣������վʾ��ͼԴ�Ի�������ͼƬ�����ο�������ʵ�ʲ��Խ��Ϊ��������Ȩ����ϵ��������ɾ������Ʒ���������ο�������ʵ��ֵΪ�� |
|
��Ȩ���� © 2019 ���������²ĿƼ�����˾
All rights reserved. ��ICP��16054715��-2 |
ɨһɨ