
��ѯ���ߣ�
17715390137
18101240246
18914047343
�ʼ���mxenes@163.com
ɨ���ע�����������ںţ�
������Fronrier
��ע�������½���ϵ���ǣ�
������ҵ�š�
רҵ��������


��һ���ߣ�Grigorii Skorupskii
ͨѶ���ߣ�MirceaDincă
ͨѶ��λ����ʡ����ѧԺ
�о����㣺
1. ������һϵ�л�����ϵԪ�صĶ�άMOFs�����ֶ�ά��Ƭ�Ĵ�ֱ�����Ͽ��Է�����Ч�ĵ�ɴ��䡣
2. �������ڲ�״MOFs�У��ߵ絼�ʲ���һ����Ҫ���и߹������ʵĽ�����������л�����֮�������ÿ��Բ�����Ч�ĵ�ɴ���ͨ·��
����MOFs
����������������л��Ǽ�(MOFs)��Ϊ������������ء��ȵ�װ�á���ѧ��������������������DZ�ڲ��϶��ܵ����ǵĹ㷺��ע���ڿ����絼��Խ��Խ�ߵ�MOFs����Ͷ���˴�����Ŭ����
�������£�����λ�ۺ���ĵ�ǰ��¼ֵΪ2500 S cm-1�����MOFs�ļ�¼ֵΪ40 S cm-1�����кܶ�ӽ���Щ��¼ֵ�IJ��ϣ������ѱ��������Ƿֲ��ܣ���������Ni2+��Cu2+�ȵ�һ�Ŷ��۹��ɽ����������ӵ���ȡ���ı�����������ͱ�Ϊ�����ġ���Щ���ϵĸߵ絼��ֵ�����������������-ϵͳ�ͽ���d���֮��ǿ�ҵ�ƽ�湲�
����Ŀǰ����û�й��ڵ�������-�л���Ĥ�ı�������о����������˳����ý���-��������Ƴɾ��в�ͬ�̶ȹ��ۼ��Ķ�ά(2D) MOFs������Իش�����Щ�����е�������йصĻ������⣬��������ṹ��������ʵ�ϸ����δ�б�����
��ϵMOFs
��ϵ����������һ��dz��ʺ�ϵͳ�о��ṹ�빦��֮���ϵ�Ľ������ӡ�������̬ʱ��������(Ln3+)��5d���Ӳ��ǿյģ����еļ۵��Ӷ�λ�ڸ߶����ε���4f����ϡ���Ϊ4f���û�����Բ���ɼ�������Ln3+�����γɵ����ӻ�����Ҫ�ȹ��ɽ�����ܶࡣ
���MOF�ϳɺ�������������Ϊ��MOF�ϳ��У������ȶ��ġ�����ļ��γɸ���ľ�����ϣ������ʵ�ֶԲ��ϵ��ӽṹ��ϵͳ����������Ҫ��ʹ��Ln3+����Ϊ����MOFs�İ������ӵ���һ���ô��ǣ����ǵ������Ե����˼�����ͬ�Ļ�ѧ��Ϊ������֮�⣬���ǵ����Ӱ뾶�dz���ͬ����La3+��Lu3+������15%���ϡ�
��Щ����ʹ���dz�Ϊ�о��ṹ��������Ե����빤�ߣ���Ϊ���۱�������ṹ�����ء�
�ɹ�����
�м��ڴˣ���ʡ����ѧԺ��Mircea Dincă��������ˣ���������Ln3+��2,3,6,7,10,11-���ǻ�����(H6HHTP)��ɵ�MOFs���塣����Ln3+��������������֮������Ӽ����ÿ��ܻή�Ͷ�ά2Dƽ���ڵ��ת�Ƶ�Ч�ʣ��������ϲ���˫̽��ྦྷ��������ʱ���絼��ֵ��Ȼ�ﵽ��0.05 S cm-1����Ŀǰ�������ĵ�����õ�MOFs�絼��ֵ�൱�����ϵĸ߽ᾧ��ʹ���ܹ�ϵͳ�ؿ��ƽṹ����������ʾ�˸�������4fϵ�е����ֲ�ͬ��ϵ���ϵ�MOFs�Ķѻ����롢�絼�ʺ�ѧ��϶֮���ֱ�ӹ�ϵ��
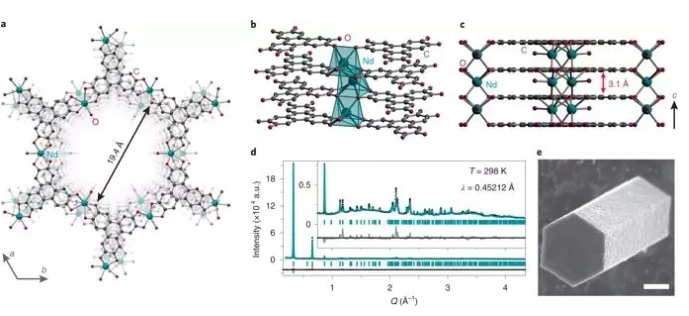
ͼ1. ��ϵMOFs�ṹ��
Ҫ��1���ϳɺͽṹ����
H6HHTP��ˮ��Ln(NO3)3 (Ln = La, Nd, Ho, Yb)�Ļ����Һ���ٽ��N,N-����������ͪ(DMI)�����ܼ��ȷ�Ӧ���õ�Ln1+ xHHTP(H2O)n (x = 0~ 0.2;���LnHHTP)��
ɨ���������(SEM)��ʾ����ĩ����״���õ������������(ͼ1e)�����ݾ���ĺϳ��������䳤����1��200��֮��仯��ͨ��Rietveld�Է�ĩx��������(PXRD)���ݽ���ϸ�����õ������ǵĽṹ��ΪNd3+(ͼ1)��Yb3+�������ṩ�����õĽṹģ�͡������ֲ����ǵȽṹ�ģ�����Yb3+�ijߴ��Nd3+С�������ڵ�λ��Ԫ������Ln-O�����ϴ��ڲ��졣
�����ֽṹ�����ֳ�����λ��������ԣ�ÿһ�ֽṹ�����ֳ�����ͬ��ռ�ݵ�����ͽ���λ�㡣Ϊ�����������ͼ1ֻ��ʾ��ƽ���ġ������NdHHTP�ṹ��һ�롣�뱨���Ĺ��ɽ������������ƣ�Ln3+���ӽ������ε�HHTP�����ϳɾ��п��ܿķ���״��ά��״�ṹ��NdHHTP�ľ���ֱ��ԼΪ1.94 nm��ͨ�����77 Kʱ��N2���������ߣ����ֲ��ϵ�ʵ�����Ϊ~1.6 nm����ᾧѧֵһ�¡�
���ɽ������л�������ͬһƽ���ϣ��γ��ϸ�Ķ�ά(2D)ƽ�棬�������Ln3+����λ���л������ƽ��֮�䣬�Ӷ����������ӳ�һ����ά���硣��ϵԪ�ر������ڽ������е�6����ԭ�Ӻ�1��ˮ�����������������DZ��Žӳ������������Ա�ʾΪ�߹�����ñ״���������������Nd2O3���ڵ�ϡ�����������λ��������չ�ṹ���ơ�������NdHHTP����YbHHTP����ϵԪ�ص�λ�ö�û�б���ȫռ�ݣ�����������֮һ��λ���ǿյģ����Ǹ��ݹ�ʽ����Ľ����
Ҫ��2�������ܴ��ṹ����
�ӽ��Ħ�-�ѻ��ٽ���Ч�������ھ���c�ķ������ص����Ӷ��ٽ���ɴ���������ά��Ƭ���о����Dz����ܶȷ�������(DFT) ������ӽṹ���Ͻ����˵��顣Ϊ�˱�����������ȴ���ļ����ܼ��������������㣬�о����ǶԷ�տDz��LaHHTP�ṹ�ͼ����LuHHTP�ṹ�����˼��㣬������Ϊ��С��ϵԪ��Yb3+��Ho3+��ģ�͡�
��Щ�����������������c����Ӧ�ñ��ֳ�������Ϊ (��ͼ2�в���Ԩ������A����)���Ӵ�Խ�����ܼ����ܴ����Կ��������֮�£����ܴ��з����ܼ�λ��a-bƽ��(��-K-M)����ƽ�����л����ƽ����(��-K-M)���ֳ��뵼����Ϊ������Ԥ�ڵ���������ϵԪ�صļ۲����Է����ܼ���Χ���ܴ����ײ������������֮���ƽ���ڵ���ͨ�ź��١�
��ʵ�ϣ��ܴ����Ŧ�-K-M������������ƽ̹�ģ��γɵġ����ڡ���϶(��Χ��1.2 ~ 0.7 eV)ǿ�������ڶѻ����롣��֮�γ������Աȵ��ǣ��ܴ��ṹ����A��������(�γɡ�����ƽ�桱)���ܴ�������ۼ����ԣ������˵͵�̬�ܶ�(DOS)�����⣬��ͬ����ԭ�ӶԽṹ�Ľ�ģ���´�La��Lu�ı仯�����ԡ�
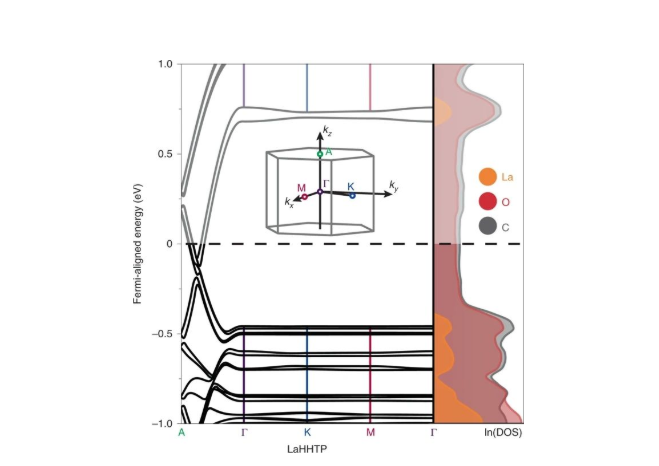
ͼ2. LaHHTP�ĵ����ܴ��ṹ��DOSͼ��
Ҫ��3�������о�
���ֲ��ϵ����������(ͼ3a)��0.7-1eV��Χ����ʾ�����������ձ�Ե���о����ǽ�����ΪDFT����Ԥ������ڴ�϶���ĸ�MOFs�ڴ˱�Ե����Ҳ���ֳ�����������La��NdԼ0.3 eV, Ho��YbԼ0.7 eV��Ȼ������Щ����������ֱ�ӻ��Ӵ�϶��Tauc�����ϻ���ʱ����������������˿��Թ�����ȱ�ݵ����ա�
��[Ru3HHTP]2+��������2000-3000 nm�����������������������ڵ��ת�ơ���Tauc�����л��Ƶ�ֱ������ԾǨ(ͼ3b)��������ʾ��һ�����˾��ȵ����ƣ����ӽϴ����ϵԪ��LaHHTP��0.85 eV����С����ϵԪ��YbHHTP��0.73 eV��ԾǨ��������С�˹�ѧ��϶��
��һ��������������뵼��(����GaAs��InP��Ge)���۲쵽�������෴������Щ�뵼���У���ѹ�ͽ��͵�Ԫ��Ԫ�����ᵼ�´�϶����Ȼ����LnHHTP���ϵ��������ڷ�������ת��PbE (E = S, Se, Te)�Ȱ뵼���Լ�����Si���ڵ�һЩ�����϶�뵼���й۲쵽�����һ�¡�����һ���Ƶľ�ȷ��λ��Ҫ��һ����ϵͳʵ�顣
Ȼ�����о�����ע��������DFT���㽫�����ܶ�λ��HHTP3-�����������ϣ���ʵ�鹫ʽ��������������̬���Ÿ���Ļ�ԭ�������仯���⽫ʹ�����ܱ�ø��ߡ�
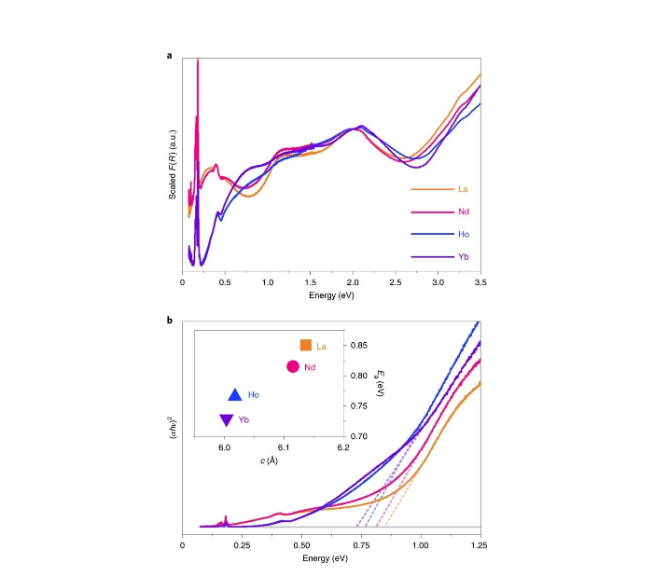
ͼ3. LnHHTP�����������(Ln = La, Nd, Ho,Yb)��
Ҫ��4���絼��
��LnHHTP�����ƳɵĶྦྷ��ĵ絼��������Ϊֹ��������õĶ��MOFs�൱����LaHHTP��0.9��10-4 S cm-1��HoHHTP��0.05 S cm-1 (ͼ4a)������ÿһ�ֲ��ϵ�������֮��IJ�����Խϴ����ڽ�С����ϵԪ��(Ho,Yb)�����Ի�ø��ߵ�ƽ�����ߵ������絼�ʣ�����������ЩԪ�ز����˸�Ϊ�ܼ��Ķѻ��ṹ��
ֵ��ע����ǣ�Ln-O����LnHHTP�����й��۽ϵ͡���Ҫ���ǣ����ڵ�ɴ����DZ�ڸ��������Լ����ӵĽӴ��;�����裬����LnHHTP MOFs��ֵ���ܱ����ص�����Ȼ��Щ���������������������е�MOFs�����ֳ����Ƶ���ò�;����ߴ磬������DZ��ٶ�Ϊ�Բ�ͬ���Ͼ���һ���������ԡ���˫̽������ȣ�����MOFs�ĵ絼��������������������ϡ�
���µ絼�ʲ��������ʾ�����ֲ��ϵ��ȼ�������(ͼ4b)����������˹������ϵ絼���¶�֮�乫ʽΪG = G0exp (EA/kT)������G�ǵ絼,G0ǰ����,EA�ǻ�ܣ�k��������������T�¶ȣ���225-300k�¶ȷ�Χ�ڣ����������ĸ����ϣ���ʾ���Ƶ�EA����ֵ��ԼΪ0.25 eV����Щֵ�������ߵ�����MOFs�ı���ֵһ�£�������ѧ��ص�2,5-���ǻ��������ȱ�������������ϡ�
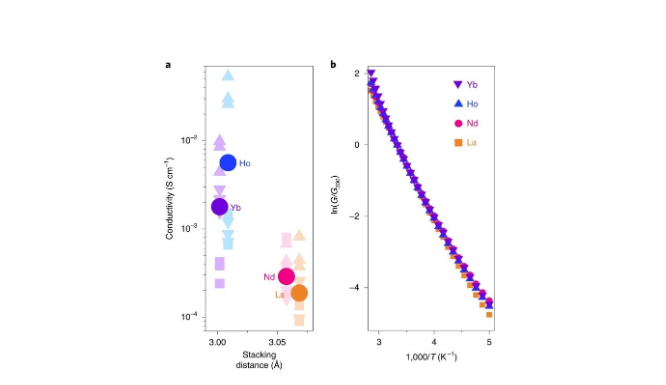
ͼ4. LnHHTP�ĵ絼��(Ln = Yb, Ho, Nd, La)��
��
Ŀǰ��2D���ϵ���Ȥ��Ҫ������ƽ���ڵĵ��������ϣ�����ƽ�����������ʵĹ�ע��Խ��١��봫ͳ����2D������ȣ�MOFs����ͨ�������Ӳ������ܲ��ϣ���Щ���ܲ��ϵ�2D�����֮��ͨ��ǿ�����ӡ�
�о�����֤������ϵ�����봫ͳ�����ںϳɶ�άMOFs�����巴Ӧ���Եõ���״���ϣ������л������γ��˱�Ƭ����ƽ���ڵĵ���ͨ�Ų�����Ҫ����������Ҫ�����ڴ�ֱ��ƽ��ķ����ϣ����ܵ����и߶ȿɵ��ԵIJ��ѻ������Ӱ�죬�����ѻ�����������ϵ�����ӵİ뾶��С�ɱ����ر仯����Щ���Ϊ�ɵ��ԵĶ�άMOFs�д���Ĺ淶�����ṩ����һ�ֹ۵㣬�ý��ͼ�����ȫ������ƽ������ϡ����о��ķ�����������Щ��ײ����п��ܲ�����Ч���������õķ�Χ���Ӷ�Ϊ���м�¼�絼�ʺ���������ά���ϵĵ�������MOFs�ṩ�˶������Ʋ��ԡ�
�ο����ף�
Skorupskii, G., Trump, B.A., Kasel, T.W. et al. Efficientand tunable one-dimensional charge transport in layered lanthanidemetal�Corganic frameworks. Nat. Chem. 12, 131�C136 (2020).
DOI��10.1038/s41557-019-0372-0
https://doi.org/10.1038/s41557-019-0372-0
 |
 |
 |
 |
| ��ά����Frontier | �������ײ���ǰ�� | MXenes Frontier | ����ҽѧFrontier |

| ��ܰ��ʾ�����������²ĿƼ�����Ӧ��Ʒ�����ڿ��У������������塣������վʾ��ͼԴ�Ի�������ͼƬ�����ο�������ʵ�ʲ��Խ��Ϊ��������Ȩ����ϵ��������ɾ������Ʒ���������ο�������ʵ��ֵΪ�� |
|
��Ȩ���� © 2019 ���������²ĿƼ�����˾
All rights reserved. ��ICP��16054715��-2 |
ɨһɨ