 ѧ��ǰ��
ѧ��ǰ��
Advanced Materials | ����Դ��������̵�����ˮ����ִ������ʵ��˫����̬���κ�ҩ���ͷ�
QQѧ������Ⱥ��1092348845
��ϸ����
����ܹ�������Ӧ�������������������ϣ���Ȼ������ҽѧϵͳ�����е�һ�������ս�����Ľ�����һ�� 3D ��ӡ�Ļ�ϵ���-�ۺ���ˮ����ִ��������ִ����ͨ����Դ��������������ʵ������θģ�����ʵ����ȫ������˫����״���κ�ø����ҩ���ͷš�ִ������˫��ṹ��ɣ�һ�����ţѪ����ۣ��Ҷ���������������BSA-PEGDA���������㣬��һ��Ϊ���� PEGDA �㡣������θҺ�У�BSA-PEGDA �㾭�����ٹ������ͣ������θ����ø�鵼�Ľ����ӳ���������������������״ת�䣬�����˹���Ԥ��ͨ����������ǣ�DOX��Ƕ�� BSA-PEGDA ˮ���������У���ϵͳʵ�����ض���λ��ø�ſ�ҩ���ͷţ�����ͨ���� A ��Ϊ�������Ƽ����е��ڡ��߷ֱ������ֹ����DLP����ӡʹ�����츴�ӵ�����ִ�������䱸��ļг�����Ϊ���ܣ���Щ�ճ����ܹ�ʵ���Ĥճ�������ͷ���Ϊ�Լ��ܿش��ݡ����о�ȷ����һ�ֲ�����Ʋ��ԣ�������������Ϊ�ɱ������������е�����������Ϊ������Ӧ�������˺�ԭλҩ������ṩ�˼�ʵƽ̨��
 ��
��
���о�����Ϊ��Autonomous Hydrogel Actuators Programmed by Endogenous Biochemical Logic for Dual�\Stage Morphing and Drug Release��������Advanced Materials�ϡ�
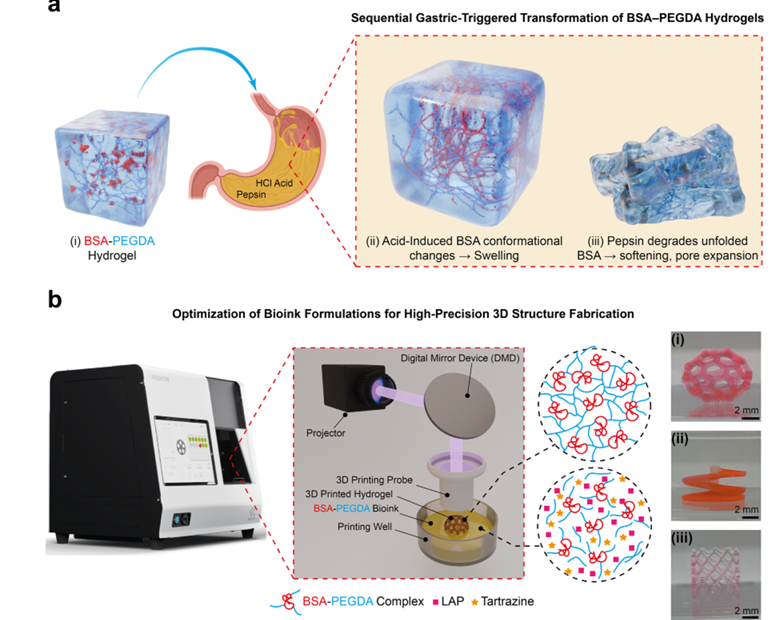
BSA-PEGDAˮ���������ø����ת�估�Ż�BSA-PEGDA����īˮ�ĸ߱���3D��ӡ��(a) BSA-PEGDAˮ������θҺ����Ӧ���Ƶ�ʾ��ͼ��(i) BSA���ӹ���Ƕ��PEGDA�����У��γɵ�һ�Ļ�Ͻṹ�������Ի����£�BSA����ɫ����ˮ�����ڱ�������״����(ii�Ciii) ��θҺ�����£�ˮ��������������������̬ת�䡣�����ڽΣ�����pH����BSA����չ�����Ժ�ɫ��������ʾ������θ����ø�鵼�Ľ�����Ȼ���٣����³�ʼ���͡�����ʱ������ƣ�θ����ø�Ľ���������ռ������λ������Խ��Խ���BSA�����⣬ˮ��������������϶����ͨ����ɫBSA�����ʧ�����粿��̮�����۲쵽��(b) BSA-PEGDAˮ�����ĺϳ���3D��ӡ��BSA-PEGDA������ͨ��Aza�CMichael�ӳɷ�Ӧ�ϳɣ�����BSA������л���һ��������PEGDA�ı�ϩ��������Ӧ�γ��ȶ��Ĺ��ۼ�������BSA-PEGDA�������LAP�����տ����أ�Tartrazine���Ʊ��ɿɹ�̻�����īˮ��Ȼ��īˮװ���䱸405 nm�Ϲ�Դ����߿���7.5 nm����BIONOVA X��CELLINK��DLP 3D�����ӡ���С���ӡʱ��ǿԼΪ12 mW cm−2����Ӧ��ӡ���ɵ���Χ4�C16 mW cm−2��75%�������Ϊ50 µm��XY�ֱ���Ϊ10 µm��Z����Ϊ4 µm�������������ÿ���ع�ʱ��Ϊ10�롣����Щ�����£�405 nm�����ܸ�Ч����LAP������PEGDA��ϩ�������ɻ��鵼�ۺϣ�ʵ�־��и߱����ˮ������㹹������ӡ����Ʒ��TRIS��Һ�г��׳�ϴ��10���ӡ�3�֣�����ȥ��δ��Ӧ�ĵ���Ͳ������������(i�Ciii) չʾ��ʹ���Ż�BSA-PEGDA����īˮ�䷽��BSA-PEGDA:LAP:���տ�����=24:6:1��v/v/v��ͨ��3D��ӡ�Ʊ�����������Ŀ���C60��֧�ܺ������ṹ����Щ�ṹ��ʾ����ȷ����̬����߽ṹ�����ԣ�ͻ�������տ���������߸��Ӽ�����ֱ����еĹؼ����á�
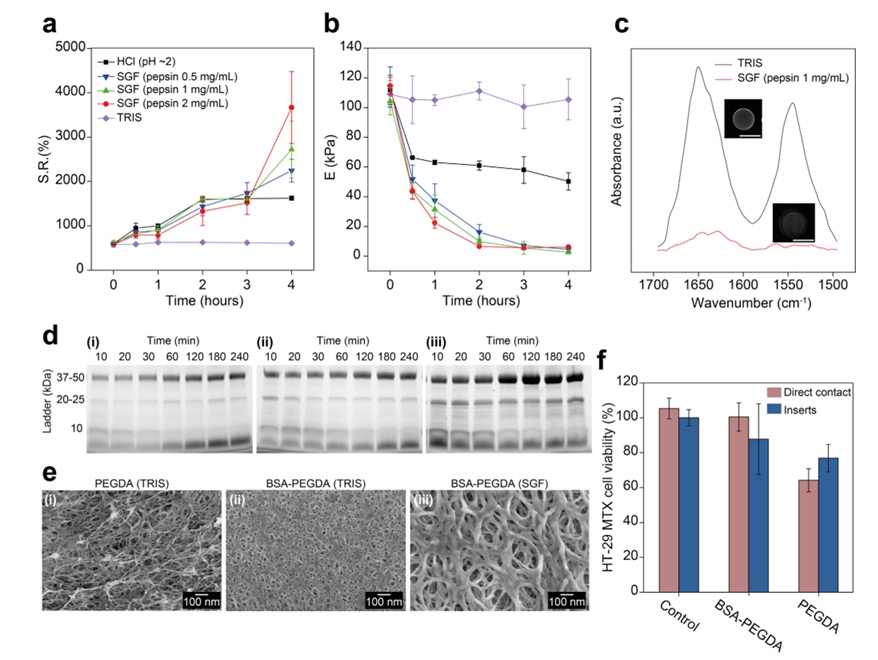
BSA-PEGDAˮ������θ��ģ����е����͡����⡢�ṹ���������������ԡ���a�� BSA-PEGDAˮ������37��C��4Сʱ����TRIS��HCl��pH ∼2����SGF��θ����ø0.5��1�� 2 mg/mL��pH ∼2���е����ͱȣ�SR���ٷֱȣ����������У�SR����2Сʱ���ȶ�����SGF�У��ΰ��ᣨ���ᣩ��SR���������������ڽ����ᣨpH ∼2��Ϊ�ͣ���Ϊθ����ø����BSA������ˮ�����ա��ϸߵ�θ����øŨ�Ȼή�ͳ������ͣ�������ˮ������ʴ��4Сʱ��SR��ﵽ��ߡ���TRIS�У�SR�����ȶ�����b�� BSA-PEGDAˮ������37��C��ѹ������ģ����E�� kPa����TRIS��HCl��pH ∼2����SGF��θ����ø0.5�� 1�� 2 mg/mL�� pH ∼2���б��֡�30����ʱ��ѹ�����ģ����SGF��HCl��pH ∼2���������½��������ᣨpH ∼2�����½��ٶ�Ҳ��SGF������SGF�У�θ����øЭͬ���ٸն���ʧ���ϸߵ�θ����øˮƽ�����˸նȵ��½�������ʱ�����ƣ�HCl��pH ∼2���ն��½��ٶȼ������ȶ���Լ60 kPa����SGF�е�E��4h���½�Լ10kPa��TRIS������ʵ���б����ȶ�Eֵ����c����37��C��θ����ø1 mg/mL��pH ∼2���У���BSA-PEGDAˮ������BSA�ṹ����ATR FTIR������������4Сʱ����TRIS����ȣ�����I�壨Լ1650 cm−1��������II�壨Լ1550 cm−1���������٣�����ø�����ƻ���BSA�Ķ����ṹ��ANS��Ϊ��ˮӫ��̽�룬��BSA����ˮ�����ϣ��Ӷ��ܹ������ؼ�⵰���ʹ���仯�ͽ��⡣�����ANSӫ��ͼ����ʾθ����øǿ�Ƚ��ͣ�������ˮ����¶���٣���һ��֤ʵ��BSA��ˮ���������еĽ��⡣�����ߣ�8���ס���d�� SDS-PAGE�����������37��C��4Сʱ����SGF��BSA�Ľ����ˮ�����ͷ����������BSA��ǿ�ȣ�37�C50 kDa��<10 kDa����ʱ�����������ø���������Ĵ�ˮ�������������ͷš���e�� TRIS��PEGDA��200 mM����BSA-PEGDA��2�C100 mM��ˮ�������䶳SEM��SGF������ø1 mg/mL��pH ∼2����37��C��4Сʱ�Ĵ�����BSA-PEGDA�γɡ�PEGDA��TRIS�����ֳ������Ҵ��϶�ṹ;BSA-PEGDA��TRIS����ʾ���ȵĿ�϶�ȡ���SGF�У�BSA-PEGDA���⣬��϶����������ɢ�Ҳ������𡣣�f�� ���÷����Һ���˳���Ƥϸ��ϵHT29-MTX����ֱ�Ӻͼ�ӣ������룩�Ӵ�����������7���BSA-PEGDA��2�C100 mM����PEGDA��200 mM��ˮ��������ϸ��������������ͨ��MTT�ⶨ��������ϸ������ʡ�BSA-PEGDA��PEGDAˮ���������ֳ���ϸ�������ԣ��ֱ𱣳�ϸ������ʳ���80%��Լ70%��֤ʵ�����������Լ��ѣ���ͼS3�������в�������n=3����������½���
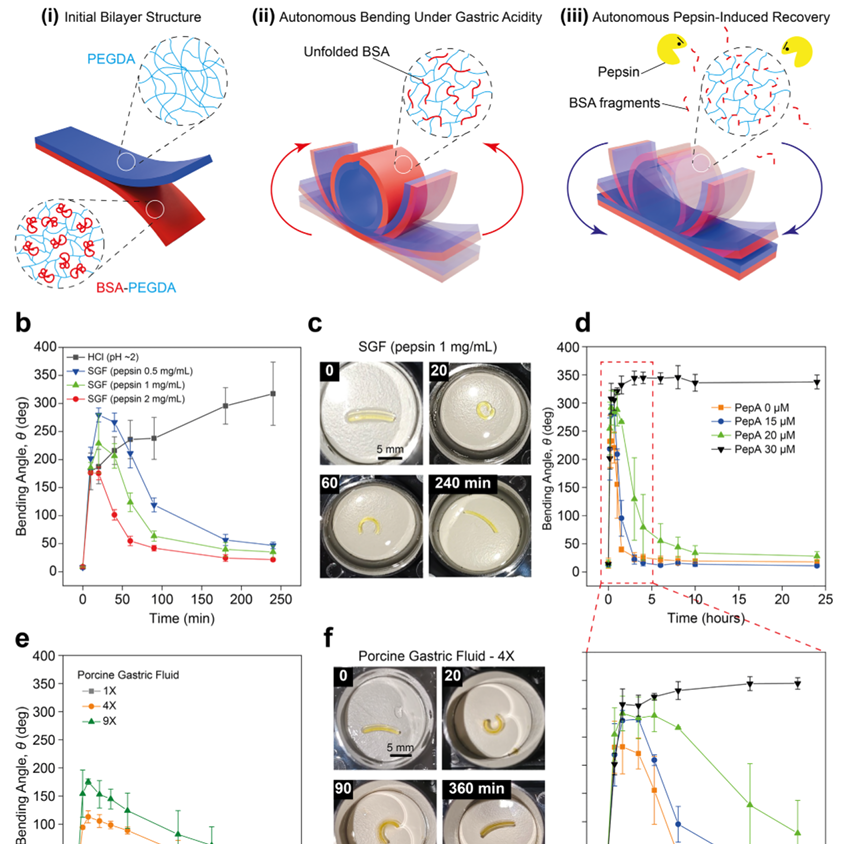
�����ԡ�ø�ٺ����Ƽ����ص�θ������˫��ˮ����ִ������˳����״���Ρ�(a) 3D��ӡ��BSA-PEGDA (2�C100 mM)/PEGDA (200 mM) ˫��ˮ����ִ����������״����ʾ��ͼ������ͨ�����۽��(i)����37◦C��SGF�У���ʼ����������BSA�Ľṹ������ǿ����ˮ�ԣ�����BSA-PEGDA������(ii)������ʱ������ƣ�BSA��ø�ٽ���������BSA-PEGDA�㣬ʹPEGDA��ָ�ԭʼ��״(iii)��(b,c) ˮ������HCl (pH ∼2)��SGFs (θ����ø0.5��1��2 mg/mL, pH ∼2)�е���״������Ӧ������ˮ����ִ������ǰ20�����������Ƕ�Ѹ�����ӣ�HCl (pH ∼2)�������³�������������4Сʱ��ﵽԼ��max ∼320◦�����֮�£���θ����ø�����£������Ƕ�������ӵ���С�����ջص�ֱ����״��SGFs�г�ʼ��������Ƕ��ڸ�Ũ��θ����ø�½ϵͣ���������BSA�����������ˮ��
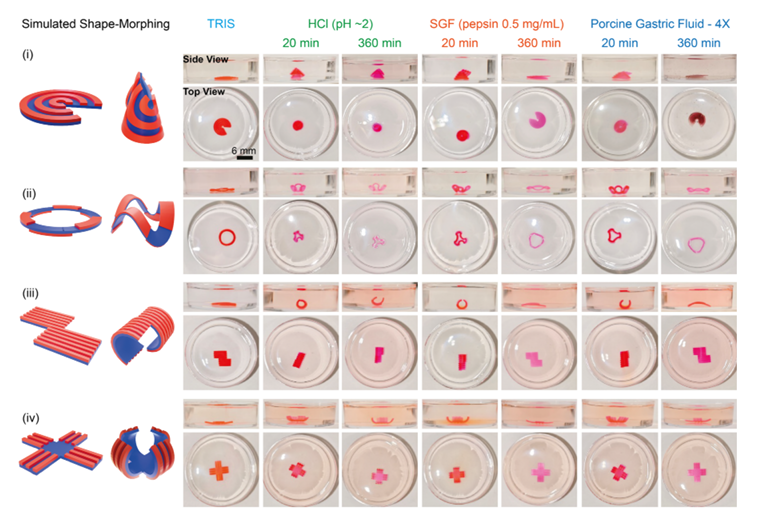
θ�ź������ĸ�����ά��ӡ˫��ˮ����ִ������״�任�����������ֲ�ͬ����ά��ӡˮ����ִ������״��i-��������ii-����iii-���ӣ�iv-���ӣ���37��C��6Сʱ���ڲ�ͬ�����е�������״�仯������HCl��pHԼ2����SGF��θ����ø 0.5 mg/mL��pHԼ2����PFG��4��Ũ�ȣ�pHԼ2������TRIS�У�ˮ����������ԭʼ��״����HCl��pHԼ2���У�ˮ������ƽ����״ת��Ϊ�ȶ�����ά�ṹ����SGF��PGF�У�ˮ����ִ���������ƽ��ṹ����Ϊ��ά�ṹ��Ȼ���ָ�����ԭʼ��״��
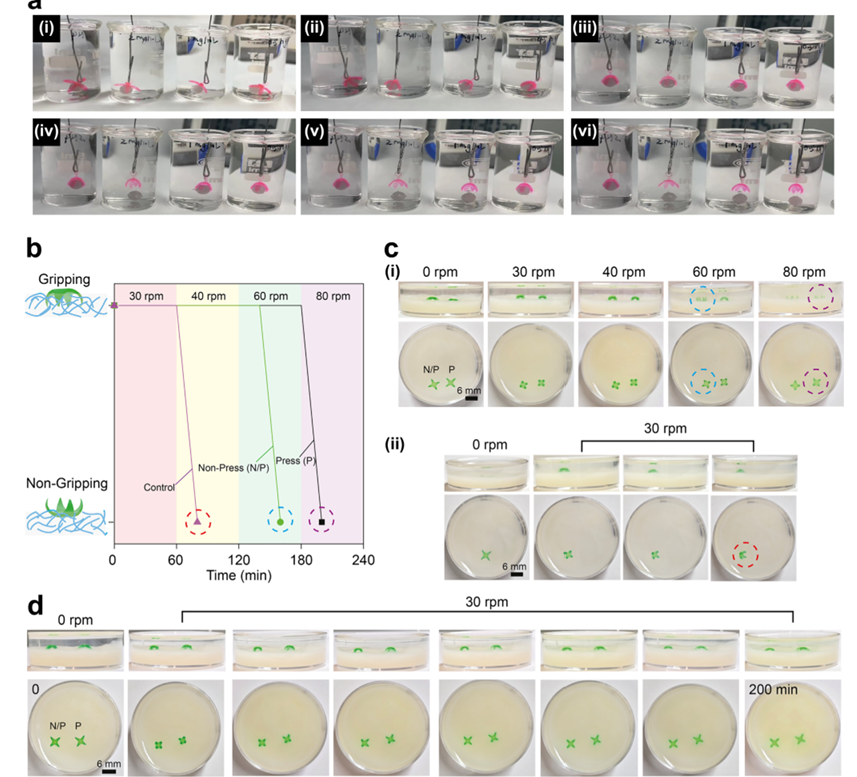
3D��ӡˮ������ǯ��ģ��θ���Һ�����е��������ͷ���Ϊ����a�� ��37��C�����£�3D��ӡˮ�����г��������ᣨpH ∼2����SGF��θ����ø2��1��0.5 mg/mL��pH ∼2����i-vi����ʵʱ��̬���Ρ�����ʱ�����ƣ���SGF�У�ˮ��������������ס������Ȼ���ɿ����ָ�ԭ�Ρ����ߵ�θ����øŨ�ȼӿ���ˮ�����Ľ��⣬����������ǰ�ͷţ���Ũ��Խ�ͣ��ͷ��ٶ�Խ�������֮�£������ᣨpH ∼2���У�ˮ��������ץ����������ɿ�����b��c����N/P�����ң�P���о������Ժ������Һ�Ļ����е�ץ�����ܡ���i�� �����ᣨpH ∼2���У�����ץ�ֶ��ܱ���30��40ת/���ӵ�ץ����;��ࣨN/P���г�����60ת/��ʱʧȥץ���������ҲࣨP���г�������80ת/����ʱʧȥץ��������ii�� �������У��г�������Ƕ��ʽ������Ȼ������ת��40ת/����ʱʧȥץ����������ץ�����ܽ�������d�� ��SGF��θ����ø1 mg/mL��pH ∼2��30ת/����ʱ���г����Զ������ͷ�ģ����Һ�㡣
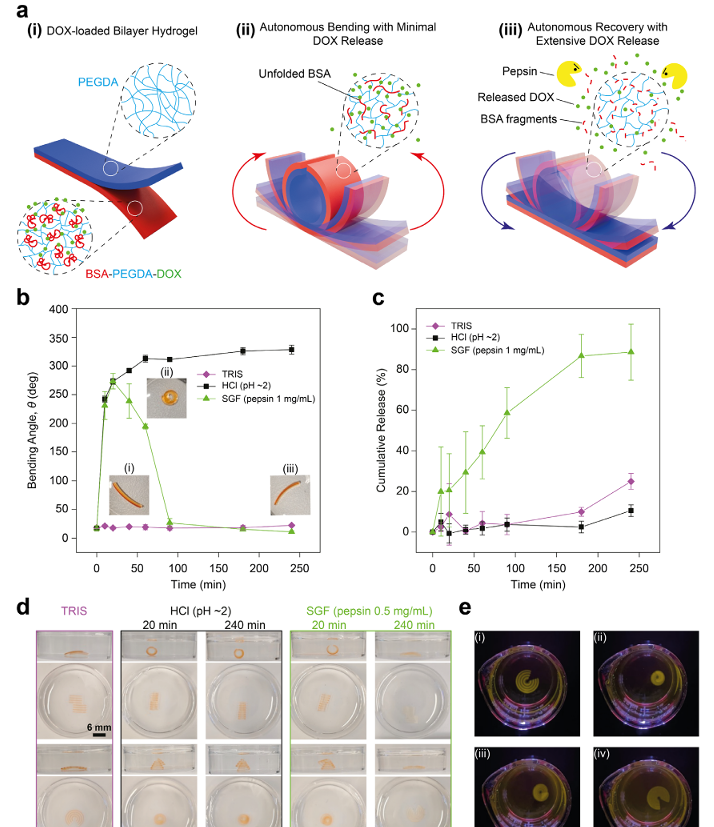
������ø���ص���״���μ�ҩ���ͷţ�������3D��ӡ��BSA-PEGDA-DOX˫��ˮ����ִ��������a�� BSA-PEGDA-DOX˫��ˮ����ִ������������̬���μ�ҩ���ͷ���Ϊ��ʾ��ͼ����i�������ˮ������PEGDA�������DOX���ص�BSA-PEGDA���Բ���ɡ���ii��������θҺ��pH ∼2���У�BSAչ�����յ�����������DOX�ͷż��١���iii������θ����ø�鵼����ļӾ磬BSA����ø�ٽ��⣬������̬�ָ��ʹ���DOX�ͷš���b�� �ڲ�ͬ�����ж�BSA-PEGDA-DOX��CDOX-����∼0.8 mg/mL��˫��ˮ��������̬���κ�DOX�ͷš�ˮ���������ᣨpH ∼2����20������Ѹ�����Ͳ���������SGF��θ����ø1mg/mL��pH∼2���У��������������60���Ӻ�ָ�ԭ״�����֮�£�TRIS��û�з�����������̬�仯����c�� SGF��θ����ø1 mg/mL��pH ∼2��������������ˮ���������DOX�ͷţ���TRIS��HCl��pH∼2�������ͷŽ�������d���ڲ�ͬ�����У�������״��BSA-PEGDA-DOX��CDOX-����∼0.8 mg/mL��ˮ����ִ�����ͷ�DOX����TRIS�У�ˮ���������ȶ�����ɫ�����ڷ����������£�����ƽ��ṹת��Ϊ��ά�ṹ����û���ͷ�DOX����ɫ������SGF��θ����ø0.5mg/mL��pH∼2�������£�����ƽ̹���ɵ���ά�ṹ��Ȼ���ָֻ�ƽ̹��ͬʱ��ȫ�ͷ�DOC����������e����ִ�������Ⱦ������յ��������������θ����ø�������BSA���ɳڣ�θ����ø0.5 mg/mL��pH ∼2����ͼ����405�����Ϲ�������¼�ƣ��Լ��DOXӫ�⡣������ӫ���źŷ�ӳ��BSA-PEGDA������ø������DOX�ͷţ�����ƵS5�������в�����ʹ��n=3������������������ƽ�������SD����ʽ����
�ܽ�
���ڡ�Advanced Materials����������չʾ��һ��ǰ�ش��£�һ����������θ�����ԡ�������ˮ��������ɫ������ѧԺ���о��Ŷ����������ţѪ����ף�BSA����θ���θ����ø��˫����Ӧ���ԣ����3D��ӡ�������������һ��˫��ṹˮ��������������θ�����Ի����£������ʽṹ�仯����ˮ����Ѹ���������Σ����ץȡ���������θ����ø��һ�ѡ�����Կ�ס������⵰�������磬ʹ�����������ָ�ԭ״��ʵ�������ġ�ץ�š�ѭ��������������ȫ�����ڹ��������źſ��ƣ������κ��ⲿ��Ԥ��
����ͻ���Ե��ǣ��о��Ŷӻ�������ҩ�ﰢù�أ�DOX�����ϵ���ϵͳ�У��������α����ҩһ�廯�ġ�����ҩе����������ץȡ�Σ�ҩ�ﱻ������ס��ֻ�е�θ����ø��ʼ���������������ָ�ʱ��ҩ��Ż�����ͷš����֡��ȶ�λ������ҩ���ľ�ʱ����ƣ�������θ����װ��һ�������������ͻ�����ҩ��ʦ��Ϊθ�������İ������ƺͻ���ҩ�ṩ��ȫ�µĽ����������־��������Ӧ��������߽������������ܻ������˹ؼ�һ����
�ο����ף�
DOI: 10.1002/adma.202516809